National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R35 GM118108
United States
Citation
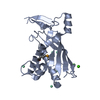
Journal: Nucleic Acids Res / Year: 2019 Title: Structural organization of a Type III-A CRISPR effector subcomplex determined by X-ray crystallography and cryo-EM. Authors: Bryan W Dorsey / Lei Huang / Alfonso Mondragón / Abstract: Clustered regularly interspaced short palindromic repeats (CRISPR) and their associated Cas proteins provide an immune-like response in many prokaryotes against extraneous nucleic acids. CRISPR-Cas ...Clustered regularly interspaced short palindromic repeats (CRISPR) and their associated Cas proteins provide an immune-like response in many prokaryotes against extraneous nucleic acids. CRISPR-Cas systems are classified into different classes and types. Class 1 CRISPR-Cas systems form multi-protein effector complexes that includes a guide RNA (crRNA) used to identify the target for destruction. Here we present crystal structures of Staphylococcus epidermidis Type III-A CRISPR subunits Csm2 and Csm3 and a 5.2 Å resolution single-particle cryo-electron microscopy (cryo-EM) reconstruction of an in vivo assembled effector subcomplex including the crRNA. The structures help to clarify the quaternary architecture of Type III-A effector complexes, and provide details on crRNA binding, target RNA binding and cleavage, and intermolecular interactions essential for effector complex assembly. The structures allow a better understanding of the organization of Type III-A CRISPR effector complexes as well as highlighting the overall similarities and differences with other Class 1 effector complexes.
Monochromator: C(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.97856 Å / Relative weight: 1
Reflection
Resolution: 2.75→77.78 Å / Num. obs: 5931 / % possible obs: 99.9 % / Redundancy: 6.6 % / CC1/2: 0.999 / Net I/σ(I): 22.7
Reflection shell
Resolution: 2.75→2.88 Å / Redundancy: 5 % / Mean I/σ(I) obs: 1.8 / Num. unique obs: 765 / CC1/2: 0.645 / % possible all: 99.8
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0232
refinement
XDS
datareduction
Aimless
datascaling
SHARP
phasing
Refinement
Method to determine structure: SAD / Resolution: 2.75→25 Å / Cor.coef. Fo:Fc: 0.939 / Cor.coef. Fo:Fc free: 0.921 / Cross valid method: THROUGHOUT / ESU R: 0.609 / ESU R Free: 0.358 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.29418
256
4.3 %
RANDOM
Rwork
0.24471
-
-
-
obs
0.24689
5635
99.71 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Staphylococcus epidermidis (bacteria)
Staphylococcus epidermidis (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj
 Assembly
Assembly

 Sample preparation
Sample preparation Processing
Processing