Transcriptional and post-translational regulation of MITF-M expression and activity / lysine-tRNA ligase / lysine-tRNA ligase activity / lysyl-tRNA aminoacylation / diadenosine tetraphosphate biosynthetic process / tRNA binding / ATP binding / cytoplasm 類似検索 - 分子機能
Bacterial/eukaryotic lysine-tRNA ligase, class II / Lysine-tRNA ligase, class II / Lysine-tRNA ligase, class II, N-terminal / Lysyl-tRNA synthetase, class II, C-terminal / Aminoacyl-tRNA synthetase, class II (D/K/N) / tRNA synthetases class II (D, K and N) / OB-fold nucleic acid binding domain, AA-tRNA synthetase-type / OB-fold nucleic acid binding domain / Bira Bifunctional Protein; Domain 2 / BirA Bifunctional Protein; domain 2 ...Bacterial/eukaryotic lysine-tRNA ligase, class II / Lysine-tRNA ligase, class II / Lysine-tRNA ligase, class II, N-terminal / Lysyl-tRNA synthetase, class II, C-terminal / Aminoacyl-tRNA synthetase, class II (D/K/N) / tRNA synthetases class II (D, K and N) / OB-fold nucleic acid binding domain, AA-tRNA synthetase-type / OB-fold nucleic acid binding domain / Bira Bifunctional Protein; Domain 2 / BirA Bifunctional Protein; domain 2 / Aminoacyl-tRNA synthetase, class II / Aminoacyl-transfer RNA synthetases class-II family profile. / Class II Aminoacyl-tRNA synthetase/Biotinyl protein ligase (BPL) and lipoyl protein ligase (LPL) / Nucleic acid-binding proteins / OB fold (Dihydrolipoamide Acetyltransferase, E2P) / Nucleic acid-binding, OB-fold / Beta Barrel / 2-Layer Sandwich / Mainly Beta / Alpha Beta 類似検索 - ドメイン・相同性
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 X線回折 /
X線回折 /  データ登録者
データ登録者 インド, 1件
インド, 1件  引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード











 PDBj
PDBj

 集合体
集合体

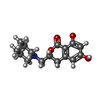
 分子量: 291.342 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C16H21NO4
分子量: 291.342 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C16H21NO4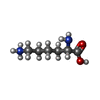
 タイプ: L-peptide linking / 分子量: 147.195 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C6H15N2O2
タイプ: L-peptide linking / 分子量: 147.195 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C6H15N2O2 分子量: 18.015 Da / 分子数: 50 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 50 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: I04 / 波長: 0.97949 Å
/ ビームライン: I04 / 波長: 0.97949 Å 解析
解析