 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6kpc | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of an agonist bound GPCR | |||||||||
 Components Components | Cannabinoid receptor 2,Endolysin | |||||||||
 Keywords Keywords | MEMBRANE PROTEIN / GPCR / LCP / agonist | |||||||||
| Function / homology |  Function and homology information Function and homology informationtrans-synaptic signaling by endocannabinoid, modulating synaptic transmission / negative regulation of mast cell activation / negative regulation of synaptic transmission, GABAergic / cannabinoid receptor activity / negative regulation of action potential / Class A/1 (Rhodopsin-like receptors) / leukocyte chemotaxis / extrinsic component of cytoplasmic side of plasma membrane / G protein-coupled receptor signaling pathway, coupled to cyclic nucleotide second messenger / viral release from host cell by cytolysis ...trans-synaptic signaling by endocannabinoid, modulating synaptic transmission / negative regulation of mast cell activation / negative regulation of synaptic transmission, GABAergic / cannabinoid receptor activity / negative regulation of action potential / Class A/1 (Rhodopsin-like receptors) / leukocyte chemotaxis / extrinsic component of cytoplasmic side of plasma membrane / G protein-coupled receptor signaling pathway, coupled to cyclic nucleotide second messenger / viral release from host cell by cytolysis / peptidoglycan catabolic process / response to amphetamine / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / adenylate cyclase-activating G protein-coupled receptor signaling pathway / response to lipopolysaccharide / G alpha (i) signalling events / host cell cytoplasm / perikaryon / postsynaptic membrane / defense response to bacterium / immune response / inflammatory response / dendrite / endoplasmic reticulum / plasma membrane / cytoplasm Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) Enterobacteria phage RB59 (virus) Enterobacteria phage RB59 (virus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.2 Å | |||||||||
 Authors Authors | Li, X.T. / Hua, T. / Wu, L.J. / Makriyannis, A. / Wu, M. / Liu, Z.J. | |||||||||
 Citation Citation | Journal: Cell / Year: 2020 Title: Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-G Complex Structures. Authors: Tian Hua / Xiaoting Li / Lijie Wu / Christos Iliopoulos-Tsoutsouvas / Yuxia Wang / Meng Wu / Ling Shen / Christina A Brust / Spyros P Nikas / Feng Song / Xiyong Song / Shuguang Yuan / ...Authors: Tian Hua / Xiaoting Li / Lijie Wu / Christos Iliopoulos-Tsoutsouvas / Yuxia Wang / Meng Wu / Ling Shen / Christina A Brust / Spyros P Nikas / Feng Song / Xiyong Song / Shuguang Yuan / Qianqian Sun / Yiran Wu / Shan Jiang / Travis W Grim / Othman Benchama / Edward L Stahl / Nikolai Zvonok / Suwen Zhao / Laura M Bohn / Alexandros Makriyannis / Zhi-Jie Liu /   Abstract: Human endocannabinoid systems modulate multiple physiological processes mainly through the activation of cannabinoid receptors CB1 and CB2. Their high sequence similarity, low agonist selectivity, ...Human endocannabinoid systems modulate multiple physiological processes mainly through the activation of cannabinoid receptors CB1 and CB2. Their high sequence similarity, low agonist selectivity, and lack of activation and G protein-coupling knowledge have hindered the development of therapeutic applications. Importantly, missing structural information has significantly held back the development of promising CB2-selective agonist drugs for treating inflammatory and neuropathic pain without the psychoactivity of CB1. Here, we report the cryoelectron microscopy structures of synthetic cannabinoid-bound CB2 and CB1 in complex with G, as well as agonist-bound CB2 crystal structure. Of important scientific and therapeutic benefit, our results reveal a diverse activation and signaling mechanism, the structural basis of CB2-selective agonists design, and the unexpected interaction of cholesterol with CB1, suggestive of its endogenous allosteric modulating role. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6kpc.cif.gz | 197.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6kpc.ent.gz | 154.8 KB | Display | PDB format |
| PDBx/mmJSON format | 6kpc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kp/6kpcftp://data.pdbj.org/pub/pdb/validation_reports/kp/6kpc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  0744C  0745C  6kpfC  6kpgC  5ztyS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 56209.934 Da / Num. of mol.: 1 / Mutation: G78L,T127A,T153L,G210A,C54T,C97A,R242E,G304E Source method: isolated from a genetically manipulated source Details: CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 ...Details: CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion,CB2, T4 lysozyme, CB2 fusion Source: (gene. exp.) Homo sapiens (human), (gene. exp.) Enterobacteria phage RB59 (virus)Gene: CNR2, CB2A, CB2B, e, RB59_126 / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P34972, UniProt: A0A097J809, lysozyme Spodoptera frugiperda (fall armyworm) / References: UniProt: P34972, UniProt: A0A097J809, lysozyme |
|---|---|
| #2: Chemical | ChemComp-E3R / 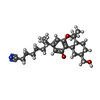  Mass: 399.566 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H37NO3 / Feature type: SUBJECT OF INVESTIGATION Mass: 399.566 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H37NO3 / Feature type: SUBJECT OF INVESTIGATION |
| Has ligand of interest | Y |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.31 Å3/Da / Density % sol: 67.19 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: lipidic cubic phase Details: 100 mM HEPES sodium pH 7.0, 25% PEG 400, 220 mM Sodium sulfate decahydrate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: Y | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL41XU / Wavelength: 1 Å / Beamline: BL41XU / Wavelength: 1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Oct 19, 2018 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 3.2→44.78 Å / Num. obs: 11476 / % possible obs: 87.6 % / Redundancy: 2.847 % / Biso Wilson estimate: 96.93 Å2 / CC1/2: 0.986 / Rmerge(I) obs: 0.171 / Rrim(I) all: 0.2 / Χ2: 0.994 / Net I/σ(I): 4.26 / Num. measured all: 32673 / Scaling rejects: 21 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Serial crystallography sample delivery | Method: fixed target |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5zty Resolution: 3.2→44.78 Å / Cor.coef. Fo:Fc: 0.905 / Cor.coef. Fo:Fc free: 0.871 / Cross valid method: THROUGHOUT / σ(F): 0 / SU Rfree Blow DPI: 0.48
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 262.08 Å2 / Biso mean: 118.21 Å2 / Biso min: 11.04 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.53 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 3.2→44.78 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.2→3.5 Å / Rfactor Rfree error: 0 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
