 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ewz: Crystal structure of RelP (SAS2) from Staphylococcus aureus bound... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ewz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of RelP (SAS2) from Staphylococcus aureus bound to AMPCPP and GTP in the pre-catalytic state | ||||||
 Components Components | GTP pyrophosphokinase | ||||||
 Keywords Keywords | TRANSFERASE / magic spot / (p)ppGpp / small alarmone synthetase / persistence / resistance / MRSA / staphylococcus | ||||||
| Function / homology |  Function and homology information Function and homology informationGTP diphosphokinase / GTP diphosphokinase activity / guanosine tetraphosphate biosynthetic process / kinase activity / GTP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.24 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.24 Å | ||||||
 Authors Authors | Manav, M.C. / Brodersen, D.E. | ||||||
| Funding support |  Denmark, 1items Denmark, 1items
| ||||||
 Citation Citation | Journal: J. Biol. Chem. / Year: 2018 Title: Structural basis for (p)ppGpp synthesis by theStaphylococcus aureussmall alarmone synthetase RelP. Authors: Manav, M.C. / Beljantseva, J. / Bojer, M.S. / Tenson, T. / Ingmer, H. / Hauryliuk, V. / Brodersen, D.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ewz.cif.gz | 195.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ewz.ent.gz | 155.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6ewz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ew/6ewzftp://data.pdbj.org/pub/pdb/validation_reports/ew/6ewz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6ex0C  5dedS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 28094.137 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria)Gene: ywaC, BER48_002454, CEJ93_03805, ERS072738_01254, ERS073583_01544, ERS074020_00750, HMPREF3211_00175 Production host: |
|---|
-Non-polymers , 5 types, 106 molecules 
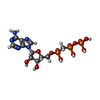



| #2: Chemical |  Mass: 523.180 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM Mass: 523.180 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM#3: Chemical |  Mass: 505.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM#4: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#5: Chemical | ChemComp-FE / |  Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 99 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.81 Å3/Da / Density % sol: 67.76 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / Details: 0.1M Tris-HCl pH8.5 25% (w/v) PEG3350 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY  / Beamline: P13 (MX1) / Wavelength: 0.979 Å / Beamline: P13 (MX1) / Wavelength: 0.979 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jun 30, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 2.24→63.07 Å / Num. obs: 42049 / % possible obs: 99.93 % / Redundancy: 13.2 % / CC1/2: 1 / Rmerge(I) obs: 0.05537 / Rpim(I) all: 0.01592 / Rrim(I) all: 0.05765 / Net I/σ(I): 24.81 |
| Reflection shell | Resolution: 2.24→2.32 Å / Redundancy: 13.1 % / Rmerge(I) obs: 1.811 / Mean I/σ(I) obs: 1.42 / CC1/2: 0.664 / Rpim(I) all: 0.5174 / Rrim(I) all: 1.885 / % possible all: 99.88 |
- Processing
Processing
| Software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5ded Resolution: 2.24→63.07 Å / Cross valid method: FREE R-VALUE
| ||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.24→63.07 Å
| ||||||||||||||||
| LS refinement shell | Resolution: 2.24→2.92 Å
|
