 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6cx0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of AtTPC1 D376A | ||||||
 Components Components | Two pore calcium channel protein 1 | ||||||
 Keywords Keywords | membrane protein/inhibitor / Ion channel / Two-pore channel / TPC1 / resting-state / closed / inactive / MEMBRANE PROTEIN / membrane protein-inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of jasmonic acid biosynthetic process / seed germination / regulation of stomatal movement / plant-type vacuole / vacuole / vacuolar membrane / monoatomic ion channel complex / voltage-gated calcium channel activity / calcium-mediated signaling / calcium channel activity ...regulation of jasmonic acid biosynthetic process / seed germination / regulation of stomatal movement / plant-type vacuole / vacuole / vacuolar membrane / monoatomic ion channel complex / voltage-gated calcium channel activity / calcium-mediated signaling / calcium channel activity / calcium ion transport / calcium ion binding / Golgi apparatus / identical protein binding / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.501 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.501 Å | ||||||
 Authors Authors | Kintzer, A.F. / Stroud, R.M. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2018 Title: Structural basis for activation of voltage sensor domains in an ion channel TPC1. Authors: Alexander F Kintzer / Evan M Green / Pawel K Dominik / Michael Bridges / Jean-Paul Armache / Dawid Deneka / Sangwoo S Kim / Wayne Hubbell / Anthony A Kossiakoff / Yifan Cheng / Robert M Stroud / Abstract: Voltage-sensing domains (VSDs) couple changes in transmembrane electrical potential to conformational changes that regulate ion conductance through a central channel. Positively charged amino acids ...Voltage-sensing domains (VSDs) couple changes in transmembrane electrical potential to conformational changes that regulate ion conductance through a central channel. Positively charged amino acids inside each sensor cooperatively respond to changes in voltage. Our previous structure of a TPC1 channel captured an example of a resting-state VSD in an intact ion channel. To generate an activated-state VSD in the same channel we removed the luminal inhibitory Ca-binding site (Ca), which shifts voltage-dependent opening to more negative voltage and activation at 0 mV. Cryo-EM reveals two coexisting structures of the VSD, an intermediate state 1 that partially closes access to the cytoplasmic side but remains occluded on the luminal side and an intermediate activated state 2 in which the cytoplasmic solvent access to the gating charges closes, while luminal access partially opens. Activation can be thought of as moving a hydrophobic insulating region of the VSD from the external side to an alternate grouping on the internal side. This effectively moves the gating charges from the inside potential to that of the outside. Activation also requires binding of Ca to a cytoplasmic site (Ca). An X-ray structure with Ca removed and a near-atomic resolution cryo-EM structure with Ca removed define how dramatic conformational changes in the cytoplasmic domains may communicate with the VSD during activation. Together four structures provide a basis for understanding the voltage-dependent transition from resting to activated state, the tuning of VSD by thermodynamic stability, and this channel's requirement of cytoplasmic Ca ions for activation. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6cx0.cif.gz | 143.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6cx0.ent.gz | 109 KB | Display | PDB format |
| PDBx/mmJSON format | 6cx0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cx/6cx0ftp://data.pdbj.org/pub/pdb/validation_reports/cx/6cx0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  8956C  8957C  8958C  8960C  6e1kC  6e1mC  6e1nC  6e1pC  5dqqS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 84310.234 Da / Num. of mol.: 1 / Mutation: D376A Source method: isolated from a genetically manipulated source Source: (gene. exp.)  | ||
|---|---|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 5 Mass: 40.078 Da / Num. of mol.: 5Source method: isolated from a genetically manipulated source Formula: Ca #3: Chemical | ChemComp-FJ7 / ( | 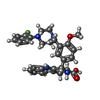  Mass: 514.591 Da / Num. of mol.: 1 Mass: 514.591 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C30H31FN4O3 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.42 Å3/Da / Density % sol: 72.2 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, sitting drop / pH: 9.3 Details: Sitting drop 2microliter drops crystchem 0.1 M glycine, pH 9.3, 50 mM potassium chloride, 1 mM calcium chloride, 35% PEG300 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 8.3.1 / Wavelength: 1 Å |
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Feb 21, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.5→30 Å / Num. obs: 19231 / % possible obs: 99.7 % / Redundancy: 13.6 % / Rrim(I) all: 0.239 / Net I/σ(I): 8.08 |
| Reflection shell | Resolution: 3.5→4 Å / Num. unique obs: 6273 / % possible all: 99.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5DQQ Resolution: 3.501→41 Å / SU ML: 0.5 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 51.07 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.501→41 Å
| ||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||
| LS refinement shell |
|
