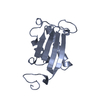
 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6c5f: Human D-Dopachrome tautomerase (D-DT)/ macrophage migration inhib... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6c5f | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human D-Dopachrome tautomerase (D-DT)/ macrophage migration inhibitory factor 2 (MIF2) complexed with the selective inhibitor 4-CPPC | ||||||
 Components Components | D-dopachrome decarboxylase | ||||||
 Keywords Keywords | CYTOKINE / selective inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationD-dopachrome decarboxylase / D-dopachrome decarboxylase activity / dopachrome isomerase activity / melanin biosynthetic process / phenylpyruvate tautomerase activity / cytokine receptor binding / negative regulation of macrophage chemotaxis / positive regulation of inflammatory response / positive regulation of tumor necrosis factor production / positive regulation of ERK1 and ERK2 cascade ...D-dopachrome decarboxylase / D-dopachrome decarboxylase activity / dopachrome isomerase activity / melanin biosynthetic process / phenylpyruvate tautomerase activity / cytokine receptor binding / negative regulation of macrophage chemotaxis / positive regulation of inflammatory response / positive regulation of tumor necrosis factor production / positive regulation of ERK1 and ERK2 cascade / : / extracellular exosome / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.4 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.4 Å | ||||||
 Authors Authors | Pantouris, G. / Lolis, E.J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2018 Title: Structural Plasticity in the C-Terminal Region of Macrophage Migration Inhibitory Factor-2 Is Associated with an Induced Fit Mechanism for a Selective Inhibitor. Authors: Pantouris, G. / Bucala, R. / Lolis, E.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6c5f.cif.gz | 159 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6c5f.ent.gz | 124 KB | Display | PDB format |
| PDBx/mmJSON format | 6c5f.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c5/6c5fftp://data.pdbj.org/pub/pdb/validation_reports/c5/6c5f | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1dptS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: _ / Beg auth comp-ID: PRO / Beg label comp-ID: PRO / Refine code: _
NCS ensembles :
|
-Components
| #1: Protein | Mass: 12593.526 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: DDT / Production host:  #2: Chemical | ChemComp-7L9 / | 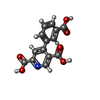  Mass: 287.224 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H9NO6 Mass: 287.224 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H9NO6#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 400 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 400 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.18 Å3/Da / Density % sol: 43.55 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.8 Details: 28% - 34% PEG 4000, 0.1 M sodium citrate tribasic dihydrate, pH 5.8, 0.2 M Ammonium acetate |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 200K / Detector: PIXEL / Date: Jan 10, 2018 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.4→50 Å / Num. obs: 63056 / % possible obs: 100 % / Redundancy: 7.9 % / Rmerge(I) obs: 0.04 / Rpim(I) all: 0.013 / Rrim(I) all: 0.042 / Χ2: 2.697 / Net I/σ(I): 33.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / % possible all: 100
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1DPT Resolution: 1.4→41.97 Å / Cor.coef. Fo:Fc: 0.979 / Cor.coef. Fo:Fc free: 0.972 / SU B: 1.431 / SU ML: 0.027 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.051 / ESU R Free: 0.05 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 73.38 Å2 / Biso mean: 15.075 Å2 / Biso min: 6.71 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.4→41.97 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Refine-ID: X-RAY DIFFRACTION / Type: interatomic distance / Weight position: 0.05
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.398→1.434 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
