 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6avk | ||||||
|---|---|---|---|---|---|---|---|
| Title | Streptavidin bound to peptide-like compound KPM-6 | ||||||
 Components Components | Streptavidin | ||||||
 Keywords Keywords | BIOTIN BINDING PROTEIN / streptavidin | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Streptomyces avidinii (bacteria) Streptomyces avidinii (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.4 Å | ||||||
 Authors Authors | Park, H. / Shamim, R. / McEnaney, P. / Kodadek, T. | ||||||
 Citation Citation | Journal: To be Published Title: Efficient Workflow for Screening DNA-encoded One Bead One Compound Libraries Using a Flow Cytometer Authors: McEnaney, P. / Dickson, P. / Dang, V. / MacConnell, A. / Cavett, V. / Reza, S. / Park, H. / Paegel, B. / Kodadek, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6avk.cif.gz | 115.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6avk.ent.gz | 88.7 KB | Display | PDB format |
| PDBx/mmJSON format | 6avk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/av/6avkftp://data.pdbj.org/pub/pdb/validation_reports/av/6avk | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1vwaS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
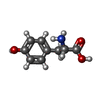
| #1: Protein | Mass: 12965.025 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Streptomyces avidinii (bacteria) / References: UniProt: P22629#2: Chemical | ChemComp-BZ4 / |   Mass: 677.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H25ClN6O10 Mass: 677.017 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H25ClN6O10#3: Chemical | ChemComp-HDO / |   Mass: 200.318 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: C12H24O2 Mass: 200.318 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: C12H24O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 302 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 302 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.79 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, sitting drop / Details: 0.2 M potassium iodide, 20% PEG3350 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.1 / Wavelength: 0.9774 Å / Beamline: 5.0.1 / Wavelength: 0.9774 Å |
| Detector | Type: DECTRIS PILATUS3 S 6M / Detector: PIXEL / Date: Aug 16, 2017 |
| Radiation | Monochromator: Si(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9774 Å / Relative weight: 1 |
| Reflection | Resolution: 1.4→69.65 Å / Num. obs: 45941 / % possible obs: 99.99 % / Redundancy: 5.8 % / Biso Wilson estimate: 16.07 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.042 / Rpim(I) all: 0.027 / Rrim(I) all: 0.05 / Net I/σ(I): 18 |
| Reflection shell | Resolution: 1.4→1.42 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.337 / Mean I/σ(I) obs: 3.4 / Num. unique all: 9975 / Num. unique obs: 2212 / CC1/2: 0.861 / Rpim(I) all: 0.239 / Rrim(I) all: 0.416 / % possible all: 98.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1VWA Resolution: 1.4→69.67 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.962 / Rfactor Rfree error: 0 / SU R Cruickshank DPI: 0.06 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.065 / SU Rfree Blow DPI: 0.061 / SU Rfree Cruickshank DPI: 0.058
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.78 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.16 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.4→69.67 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.4→1.44 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
