- PDB-6a89: Crystal structure of the ternary complex of peptidoglycan recogni... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 6a89
Title
Crystal structure of the ternary complex of peptidoglycan recognition protein (PGRP-S) with Tartaric acid, Ribose and 2,6-DIAMINOPIMELIC ACID at 2.11 A resolution
Components
Peptidoglycan recognition protein 1
Keywords
IMMUNE SYSTEM
Function / homology
Function and homology information
peptidoglycan immune receptor activity / N-acetylmuramoyl-L-alanine amidase activity / peptidoglycan binding / negative regulation of cytokine production / detection of bacterium / peptidoglycan catabolic process / defense response to Gram-positive bacterium / innate immune response / extracellular region / zinc ion binding Similarity search - Function
Peptidoglycan recognition protein, PGRP-S / Peptidoglycan recognition protein family domain, metazoa/bacteria / Peptidoglycan recognition protein / Animal peptidoglycan recognition proteins homologous to Bacteriophage T3 lysozyme. / Lysozyme-like / Peptidoglycan recognition protein-like / N-acetylmuramoyl-L-alanine amidase / Ami_2 / N-acetylmuramoyl-L-alanine amidase domain / N-acetylmuramoyl-L-alanine amidase/PGRP domain superfamily ...Peptidoglycan recognition protein, PGRP-S / Peptidoglycan recognition protein family domain, metazoa/bacteria / Peptidoglycan recognition protein / Animal peptidoglycan recognition proteins homologous to Bacteriophage T3 lysozyme. / Lysozyme-like / Peptidoglycan recognition protein-like / N-acetylmuramoyl-L-alanine amidase / Ami_2 / N-acetylmuramoyl-L-alanine amidase domain / N-acetylmuramoyl-L-alanine amidase/PGRP domain superfamily / 3-Layer(aba) Sandwich / Alpha Beta Similarity search - Domain/homology
Resolution: 2.11→86.09 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.941 / SU B: 7.829 / SU ML: 0.189 / Cross valid method: THROUGHOUT / ESU R: 0.227 / ESU R Free: 0.199 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.24884
2161
5.1 %
RANDOM
Rwork
0.19067
-
-
-
obs
0.19365
40430
99.95 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj
 Assembly
Assembly






 Type: D-saccharide, alpha linking / Mass: 150.130 Da / Num. of mol.: 1
Type: D-saccharide, alpha linking / Mass: 150.130 Da / Num. of mol.: 1


 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
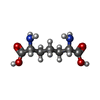
Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Type: L-peptide linking / Mass: 190.197 Da / Num. of mol.: 1
Type: L-peptide linking / Mass: 190.197 Da / Num. of mol.: 1 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3
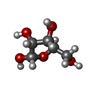
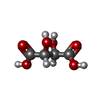
Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6
Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Sample preparation
Sample preparation / Beamline: BM14 / Wavelength: 0.9537 Å
/ Beamline: BM14 / Wavelength: 0.9537 Å Processing
Processing