sperm DNA condensation / regulation of mRNA processing / regulation of mRNA splicing, via spliceosome / Maturation of nucleoprotein / negative regulation of viral genome replication / spliceosomal complex assembly / positive regulation of viral genome replication / Replacement of protamines by nucleosomes in the male pronucleus / RNA splicing / chromosome segregation ...sperm DNA condensation / regulation of mRNA processing / regulation of mRNA splicing, via spliceosome / Maturation of nucleoprotein / negative regulation of viral genome replication / spliceosomal complex assembly / positive regulation of viral genome replication / Replacement of protamines by nucleosomes in the male pronucleus / RNA splicing / chromosome segregation / nuclear matrix / protein phosphorylation / protein kinase activity / non-specific serine/threonine protein kinase / nuclear speck / intracellular signal transduction / innate immune response / protein serine kinase activity / protein serine/threonine kinase activity / chromatin / magnesium ion binding / RNA binding / nucleoplasm / ATP binding / nucleus / plasma membrane / cytoplasm / cytosol Similarity search - Function
: / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. ...: / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
Resolution: 2.32→63.39 Å / Cor.coef. Fo:Fc: 0.937 / Cor.coef. Fo:Fc free: 0.925 / SU B: 11.98 / SU ML: 0.147 / Cross valid method: THROUGHOUT / ESU R: 0.267 / ESU R Free: 0.205 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.23797
1195
5.1 %
RANDOM
Rwork
0.21211
-
-
-
obs
0.21345
22080
99.53 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj Assembly
Assembly


 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2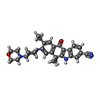
 Mass: 482.617 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C30H34N4O2
Mass: 482.617 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C30H34N4O2 Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL13B1 / Wavelength: 1 Å
/ Beamline: BL13B1 / Wavelength: 1 Å Processing
Processing