 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5wdn: Crystal structure of the influenza virus PA endonuclease in compl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5wdn | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the influenza virus PA endonuclease in complex with inhibitor 6d (SRI-29680) | ||||||||||||
 Components Components | Polymerase acidic protein | ||||||||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / Virus / Nuclease / Transcription / Cap-snatching / HYDROLASE / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationcap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / endonuclease activity / Hydrolases; Acting on ester bonds / host cell cytoplasm / symbiont-mediated suppression of host gene expression / viral translational frameshifting / viral RNA genome replication / hydrolase activity / host cell nucleus ...cap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / endonuclease activity / Hydrolases; Acting on ester bonds / host cell cytoplasm / symbiont-mediated suppression of host gene expression / viral translational frameshifting / viral RNA genome replication / hydrolase activity / host cell nucleus / DNA-templated transcription / RNA binding / metal ion binding Similarity search - Function | ||||||||||||
| Biological species |   Influenza A virus Influenza A virus | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||||||||
 Authors Authors | Kumar, G. / White, S.W. | ||||||||||||
| Funding support |  United States, 3items United States, 3items
| ||||||||||||
 Citation Citation | Journal: Sci Rep / Year: 2017 Title: Protein-Structure Assisted Optimization of 4,5-Dihydroxypyrimidine-6-Carboxamide Inhibitors of Influenza Virus Endonuclease. Authors: Beylkin, D. / Kumar, G. / Zhou, W. / Park, J. / Jeevan, T. / Lagisetti, C. / Harfoot, R. / Webby, R.J. / White, S.W. / Webb, T.R. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5wdn.cif.gz | 92.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5wdn.ent.gz | 68.8 KB | Display | PDB format |
| PDBx/mmJSON format | 5wdn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wd/5wdnftp://data.pdbj.org/pub/pdb/validation_reports/wd/5wdn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5w3iC  5w44C  5w73C  5w7uC  5w92C  5w9gC  5wa6C  5wa7C  5wapC  5wb3C  5wcsC  5wctC  5wdcC  5wdwC  5we9C  5webC  5wefC  5weiC  5wf3C  5wfmC  5wfwC  5wfzC  5wg9C C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 21970.160 Da / Num. of mol.: 1 Mutation: Loop 51-72 replaced with a GGS linker, C-Terminal has residual His-tag residues Source method: isolated from a genetically manipulated source Source: (gene. exp.) Influenza A virus / Strain: swl A/California/04/2009 H1N1 / Gene: PA / Plasmid: pET52b / Production host:  | ||||
|---|---|---|---|---|---|
| #2: Chemical |   Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn#3: Chemical | ChemComp-G1K / | 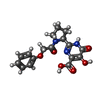  Mass: 359.333 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H17N3O6 / Feature type: SUBJECT OF INVESTIGATION Mass: 359.333 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H17N3O6 / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 45.24 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7 / Details: 0.8 M Succinic Acid pH 7.0, 2 mM MnCl2, 2 mM MgCl2 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 22-BM / Wavelength: 1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Nov 26, 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.1→50 Å / Num. obs: 12016 / % possible obs: 99.7 % / Redundancy: 10.7 % / Biso Wilson estimate: 46.27 Å2 / Rmerge(I) obs: 0.078 / Rpim(I) all: 0.025 / Rrim(I) all: 0.082 / Χ2: 1.391 / Net I/σ(I): 15.1 / Num. measured all: 129170 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.1→34.991 Å / SU ML: 0.23 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 29.25
| ||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 157.66 Å2 / Biso mean: 67.3627 Å2 / Biso min: 38.12 Å2 | ||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.1→34.991 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 5
| ||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -21.9901 Å / Origin y: 16.6764 Å / Origin z: 0.6972 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
