Resolution: 2.2→46.19 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.952 / WRfactor Rfree: 0.2443 / WRfactor Rwork: 0.2055 / FOM work R set: 0.773 / SU B: 16.027 / SU ML: 0.179 / SU R Cruickshank DPI: 0.2002 / SU Rfree: 0.1763 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.2 / ESU R Free: 0.176 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2363
678
4.7 %
RANDOM
Rwork
0.2003
-
-
-
obs
0.202
13622
99.23 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Influenza A virus
Influenza A virus X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States,
United States,  Switzerland, 3items
Switzerland, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














 PDBj
PDBj Assembly
Assembly



 Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn
Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn
 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4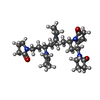
 Mass: 541.682 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C29H43N5O5
Mass: 541.682 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C29H43N5O5 Mass: 18.015 Da / Num. of mol.: 21 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 21 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing