 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5wbp: Structure of human Ketohexokinase complexed with hits from fragme... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5wbp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human Ketohexokinase complexed with hits from fragment screening | ||||||
 Components Components | Ketohexokinase | ||||||
 Keywords Keywords | TRANSFERASE / Ketohexokinase / Fragment-based drug discovery / SBDD | ||||||
| Function / homology |  Function and homology information Function and homology informationEssential fructosuria / ketohexokinase / ketohexokinase activity / fructose binding / Fructose catabolism / regulation of glycogen metabolic process / response to sucrose / response to fructose / fructose metabolic process / response to zinc ion ...Essential fructosuria / ketohexokinase / ketohexokinase activity / fructose binding / Fructose catabolism / regulation of glycogen metabolic process / response to sucrose / response to fructose / fructose metabolic process / response to zinc ion / response to glucose / response to insulin / protein homodimerization activity / extracellular exosome / ATP binding / identical protein binding / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.74 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.74 Å | ||||||
 Authors Authors | Pandit, J. | ||||||
 Citation Citation | Journal: J. Med. Chem. / Year: 2017 Title: Discovery of Fragment-Derived Small Molecules for in Vivo Inhibition of Ketohexokinase (KHK). Authors: Huard, K. / Ahn, K. / Amor, P. / Beebe, D.A. / Borzilleri, K.A. / Chrunyk, B.A. / Coffey, S.B. / Cong, Y. / Conn, E.L. / Culp, J.S. / Dowling, M.S. / Gorgoglione, M.F. / Gutierrez, J.A. / ...Authors: Huard, K. / Ahn, K. / Amor, P. / Beebe, D.A. / Borzilleri, K.A. / Chrunyk, B.A. / Coffey, S.B. / Cong, Y. / Conn, E.L. / Culp, J.S. / Dowling, M.S. / Gorgoglione, M.F. / Gutierrez, J.A. / Knafels, J.D. / Lachapelle, E.A. / Pandit, J. / Parris, K.D. / Perez, S. / Pfefferkorn, J.A. / Price, D.A. / Raymer, B. / Ross, T.T. / Shavnya, A. / Smith, A.C. / Subashi, T.A. / Tesz, G.J. / Thuma, B.A. / Tu, M. / Weaver, J.D. / Weng, Y. / Withka, J.M. / Xing, G. / Magee, T.V. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5wbp.cif.gz | 129.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5wbp.ent.gz | 98.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5wbp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wb/5wbpftp://data.pdbj.org/pub/pdb/validation_reports/wb/5wbp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5wbmC  5wboC  5wbqC  5wbrC  5wbzC  3nbvS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |


















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34076.586 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: KHK / Production host:  #2: Chemical | ChemComp-A2J / | 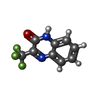  Mass: 214.144 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H5F3N2O Mass: 214.144 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H5F3N2O#3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 25 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 25 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.59 Å3/Da / Density % sol: 65.72 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 4.5 Details: 17% PEG 8k, 0.1M Na-Citrate, 0.2M Ammonium sulfate, pH 4.5 Temp details: Room temperature |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Oct 2, 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.74→138.78 Å / Num. obs: 26032 / % possible obs: 98.6 % / Redundancy: 6.1 % / Biso Wilson estimate: 95.74 Å2 / Rpim(I) all: 0.018 / Rrim(I) all: 0.046 / Rsym value: 0.038 / Net I/av σ(I): 14.2 / Net I/σ(I): 25.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 3NBV Resolution: 2.74→71.17 Å / Cor.coef. Fo:Fc: 0.9463 / Cor.coef. Fo:Fc free: 0.9313 / SU R Cruickshank DPI: 0.398 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.391 / SU Rfree Blow DPI: 0.26 / SU Rfree Cruickshank DPI: 0.265
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 174.09 Å2 / Biso mean: 89.38 Å2 / Biso min: 41.46 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.379 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.74→71.17 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.74→2.85 Å / Rfactor Rfree error: 0 / Total num. of bins used: 13
|
