Glycoside hydrolase family 11/12, catalytic domain / Glycoside hydrolase family 11, active site 2 / Glycosyl hydrolases family 11 (GH11) active site signature 2. / Glycoside hydrolase family 11, active site 1 / Glycosyl hydrolases family 11 (GH11) active site signature 1. / Glycoside hydrolase family 11 / Glycosyl hydrolases family 11 (GH11) domain / Glycosyl hydrolases family 11 / Glycosyl hydrolases family 11 (GH11) domain profile. / Glycoside hydrolase family 11/12 ...Glycoside hydrolase family 11/12, catalytic domain / Glycoside hydrolase family 11, active site 2 / Glycosyl hydrolases family 11 (GH11) active site signature 2. / Glycoside hydrolase family 11, active site 1 / Glycosyl hydrolases family 11 (GH11) active site signature 1. / Glycoside hydrolase family 11 / Glycosyl hydrolases family 11 (GH11) domain / Glycosyl hydrolases family 11 / Glycosyl hydrolases family 11 (GH11) domain profile. / Glycoside hydrolase family 11/12 / Concanavalin A-like lectin/glucanase domain superfamily / Jelly Rolls / Sandwich / Mainly Beta Similarity search - Domain/homology
Mass: 18.015 Da / Num. of mol.: 624 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.09 Å3/Da / Density % sol: 41.13 %
Crystal grow
Temperature: 293.15 K / Method: vapor diffusion, sitting drop / pH: 3.5 Details: 1ul of protein at 20mg/ml mixed with 1ul of mother liquor, plus 0.2ul of a seed stock made from a previous crystallization drop. Crystallization condition is 0.1M Citric Acid pH 3.5, 25% PEG 3350. Temp details: Temperature Controlled Crystal Incubator
-
Data collection
Diffraction
Mean temperature: 80 K / Ambient temp details: Liquid Nitrogen Cryo Stream
Diffraction source
Source: SYNCHROTRON / Site: ALS / Beamline: 8.2.2 / Wavelength: 0.75141 Å
Monochromator: Double-crystal Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.75141 Å / Relative weight: 1
Reflection
Resolution: 1→44.333 Å / Num. obs: 181340 / % possible obs: 100 % / Redundancy: 4.5 % / Biso Wilson estimate: 8.14 Å2 / Rmerge(I) obs: 0.058 / Net I/av σ(I): 21 / Net I/σ(I): 6.6
Reflection shell
Resolution (Å)
Redundancy (%)
Rmerge(I) obs
Mean I/σ(I) obs
CC1/2
Diffraction-ID
% possible all
1-1.02
3.7
0.973
0.89
0.557
1
99.9
1.02-1.04
4.1
0.793
1.22
0.681
1
100
1.04-1.06
4.4
0.682
1.53
0.754
1
100
1.06-1.08
4.5
0.528
2
0.839
1
100
1.08-1.1
4.5
0.425
2.63
0.882
1
100
1.1-1.13
4.5
0.337
3.44
0.929
1
100
1.13-1.15
4.5
0.284
4.06
0.948
1
100
1.15-1.19
4.5
0.253
4.8
0.955
1
100
1.19-1.22
4.5
0.227
5.2
0.961
1
100
1.22-1.26
4.5
0.202
6.29
0.968
1
100
1.26-1.3
4.6
0.18
6.71
0.976
1
100
1.3-1.36
4.6
0.154
8.15
0.981
1
100
1.36-1.42
4.6
0.129
9.69
0.986
1
100
1.42-1.49
4.6
0.106
12.3
0.99
1
100
1.49-1.59
4.6
0.089
16.46
0.993
1
100
1.59-1.71
4.6
0.078
20.07
0.993
1
100
1.71-1.88
4.6
0.064
26.35
0.995
1
100
1.88-2.15
4.5
0.045
36.77
0.997
1
100
2.15-2.71
4.6
0.034
41.58
0.998
1
100
2.71-50
4.7
0.023
54.12
0.999
1
100
-
Phasing
Phasing
Method: molecular replacement
Phasing MR
Highest resolution
Lowest resolution
Rotation
1.04 Å
19.29 Å
Translation
1.04 Å
19.29 Å
-
Processing
Software
Name
Version
Classification
HKL-2000
v708c
datareduction
HKL-2000
v708c
datascaling
PHASER
2.7.0
phasing
Coot
0.8.2
modelbuilding
PHENIX
dev_2841
refinement
PDB_EXTRACT
3.2
dataextraction
HKL
datareduction
HKL
datascaling
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: Fen49 Computational Design, with residues 63, 85-95 and 116-122 removed Resolution: 1→44.333 Å / SU ML: 0.08 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 10.88 Details: Interative rounds of model building in Coot and refinement in Phenix. Refinement in real and reciprocal space, all-atom (except H) anisotropic ADP refinement, occupancy refinement. ...Details: Interative rounds of model building in Coot and refinement in Phenix. Refinement in real and reciprocal space, all-atom (except H) anisotropic ADP refinement, occupancy refinement. Optimization of X-ray to stereochemistry and X-ray to ADP weights. Automatic addition of hydrogens to the model, and automatic correction of N/Q/H errors. Several rounds of updating waters during refinement. Manual inspection and correction of waters before the final round of refinement.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 3items
United States, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

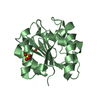



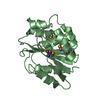





 PDBj
PDBj
 Assembly
Assembly



 Mass: 458.541 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H42O11 / Comment: precipitant*YM
Mass: 458.541 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H42O11 / Comment: precipitant*YM Mass: 18.015 Da / Num. of mol.: 624 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 624 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing