 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5noo: Crystal Structure of C.elegans Thymidylate Synthase in Complex wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5noo | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of C.elegans Thymidylate Synthase in Complex with dUMP and Tomudex | |||||||||
 Components Components | Thymidylate synthase | |||||||||
 Keywords Keywords | TRANSFERASE / enzyme / nematode / complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationthymidylate synthase / thymidylate synthase activity / dTTP biosynthetic process / dTMP biosynthetic process / methylation Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | |||||||||
 Authors Authors | Wilk, P. / Jarmula, A. / Maj, P. / Dowiercial, A. / Banaszak, K. / Rypniewski, W. / Rode, W. | |||||||||
| Funding support |  Poland, 2items Poland, 2items
| |||||||||
 Citation Citation | Journal: J. Mol. Graph. Model. / Year: 2017 Title: Crystal structures of nematode (parasitic T. spiralis and free living C. elegans), compared to mammalian, thymidylate synthases (TS). Molecular docking and molecular dynamics simulations in ...Title: Crystal structures of nematode (parasitic T. spiralis and free living C. elegans), compared to mammalian, thymidylate synthases (TS). Molecular docking and molecular dynamics simulations in search for nematode-specific inhibitors of TS. Authors: Jarmua, A. / Wilk, P. / Maj, P. / Ludwiczak, J. / Dowiercia, A. / Banaszak, K. / Rypniewski, W. / Ciesla, J. / Dabrowska, M. / Fraczyk, T. / Bronowska, A.K. / Jakowiecki, J. / Filipek, S. / Rode, W. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5noo.cif.gz | 241.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5noo.ent.gz | 194.8 KB | Display | PDB format |
| PDBx/mmJSON format | 5noo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/no/5nooftp://data.pdbj.org/pub/pdb/validation_reports/no/5noo | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4iqbC  5by6C  4eb4S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 35855.902 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-UMP / 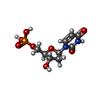  Mass: 308.182 Da / Num. of mol.: 4 Mass: 308.182 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C9H13N2O8P / References: thymidylate synthase #3: Chemical | ChemComp-D16 /   Mass: 458.488 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H22N4O6S / Comment: chemotherapy, inhibitor*YM Mass: 458.488 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H22N4O6S / Comment: chemotherapy, inhibitor*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.71 Å3/Da / Density % sol: 54.65 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / Details: 0.1M Bis-Tris pH 7.0, 0.2M NaAcetate, 17% PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SEALED TUBE / Type: OXFORD DIFFRACTION NOVA / Wavelength: 1.5418 Å |
| Detector | Type: AGILENT ATLAS CCD / Detector: CCD / Date: Dec 3, 2010 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→14.7 Å / Num. obs: 32822 / % possible obs: 96.4 % / Redundancy: 1.8 % / Rmerge(I) obs: 0.135 / Net I/σ(I): 4.6 |
| Reflection shell | Resolution: 2.9→3.06 Å / Redundancy: 2.1 % / Rmerge(I) obs: 0.375 / Num. unique all: 4929 / Rsym value: 0.375 / % possible all: 99.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4EB4 Resolution: 2.9→14.7 Å / Cross valid method: FREE R-VALUE
| |||||||||||||||||||||
| Displacement parameters | Biso max: 158.43 Å2 / Biso mean: 41.6245 Å2 / Biso min: 8.07 Å2 | |||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→14.7 Å
| |||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→2.9847 Å
|
