Formation of the polybromo-BAF (pBAF) complex / regulation of G0 to G1 transition / RSC-type complex / regulation of nucleotide-excision repair / regulation of mitotic metaphase/anaphase transition / SWI/SNF complex / positive regulation of T cell differentiation / positive regulation of double-strand break repair / nuclear chromosome / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known ...Formation of the polybromo-BAF (pBAF) complex / regulation of G0 to G1 transition / RSC-type complex / regulation of nucleotide-excision repair / regulation of mitotic metaphase/anaphase transition / SWI/SNF complex / positive regulation of T cell differentiation / positive regulation of double-strand break repair / nuclear chromosome / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / regulation of G1/S transition of mitotic cell cycle / positive regulation of myoblast differentiation / positive regulation of cell differentiation / transcription elongation by RNA polymerase II / kinetochore / nuclear matrix / RMTs methylate histone arginines / mitotic cell cycle / chromatin remodeling / negative regulation of cell population proliferation / chromatin binding / regulation of transcription by RNA polymerase II / chromatin / DNA binding / nucleoplasm / nucleus Similarity search - Function
Protein polybromo-1, Bromodomain 5 / Remodelling complex subunit Rsc/polybromo / Bromo adjacent homology domain / BAH domain / Bromo adjacent homology (BAH) domain / Bromo adjacent homology (BAH) domain superfamily / BAH domain profile. / HMG (high mobility group) box / HMG boxes A and B DNA-binding domains profile. / high mobility group ...Protein polybromo-1, Bromodomain 5 / Remodelling complex subunit Rsc/polybromo / Bromo adjacent homology domain / BAH domain / Bromo adjacent homology (BAH) domain / Bromo adjacent homology (BAH) domain superfamily / BAH domain profile. / HMG (high mobility group) box / HMG boxes A and B DNA-binding domains profile. / high mobility group / High mobility group box domain / High mobility group box domain superfamily / Bromodomain-like / Histone Acetyltransferase; Chain A / Bromodomain, conserved site / Bromodomain signature. / Bromodomain / bromo domain / Bromodomain / Bromodomain (BrD) profile. / Bromodomain-like superfamily / Up-down Bundle / Mainly Alpha Similarity search - Domain/homology
Rfactor: 43.62 / Model details: Phaser MODE: MR_AUTO
Highest resolution
Lowest resolution
Rotation
2.5 Å
23.82 Å
Translation
2.5 Å
23.82 Å
-
Processing
Software
Name
Version
Classification
SCALA
3.3.16
datascaling
PHASER
2.1.4
phasing
REFMAC
5.5.0110
refinement
PDB_EXTRACT
3.15
dataextraction
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.87→23.8 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.934 / WRfactor Rfree: 0.2411 / WRfactor Rwork: 0.1956 / FOM work R set: 0.7919 / SU B: 7.658 / SU ML: 0.112 / SU R Cruickshank DPI: 0.1547 / SU Rfree: 0.1512 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.155 / ESU R Free: 0.151 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2483
1782
4.1 %
RANDOM
Rwork
0.2007
-
-
-
obs
0.2026
41808
86.95 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj




 Assembly
Assembly





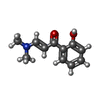
 Mass: 191.226 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C11H13NO2
Mass: 191.226 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C11H13NO2
 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 18.015 Da / Num. of mol.: 232 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 232 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing