 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
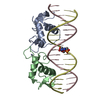
Yorodumi- PDB-5do6: Crystal structure of Dendroaspis polylepis venom mambalgin-1 T23A... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5do6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Dendroaspis polylepis venom mambalgin-1 T23A mutant | ||||||
 Components Components | Mambalgin-1 | ||||||
 Keywords Keywords | TOXIN / Acid Sensing Ion Channels / Pain suppression Drug / Elapid Venom polypeptide | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Dendroaspis polylepis polylepis (black mamba) Dendroaspis polylepis polylepis (black mamba) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.697 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.697 Å | ||||||
 Authors Authors | Stura, E.A. / Tepshi, L. / Kessler, P. / Gilles, M. / Servent, D. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2016 Title: Mambalgin-1 Pain-relieving Peptide, Stepwise Solid-phase Synthesis, Crystal Structure, and Functional Domain for Acid-sensing Ion Channel 1a Inhibition. Authors: Mourier, G. / Salinas, M. / Kessler, P. / Stura, E.A. / Leblanc, M. / Tepshi, L. / Besson, T. / Diochot, S. / Baron, A. / Douguet, D. / Lingueglia, E. / Servent, D. #1: Journal: J Synchrotron Radiat / Year: 2017 Title: Comparison of helical scan and standard rotation methods in single-crystal X-ray data collection strategies. Authors: Polsinelli, I. / Savko, M. / Rouanet-Mehouas, C. / Ciccone, L. / Nencetti, S. / Orlandini, E. / Stura, E.A. / Shepard, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5do6.cif.gz | 42.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5do6.ent.gz | 29.6 KB | Display | PDB format |
| PDBx/mmJSON format | 5do6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/do/5do6ftp://data.pdbj.org/pub/pdb/validation_reports/do/5do6 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 6544.618 Da / Num. of mol.: 2 / Fragment: UNP residues 22-78 / Mutation: T23A / Source method: obtained synthetically / Details: T23A mutant Source: (synth.) Dendroaspis polylepis polylepis (black mamba)References: UniProt: P0DKR6 #2: Chemical | ChemComp-IOD /   Mass: 126.904 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: I Mass: 126.904 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: I#3: Chemical | ChemComp-EDO / |   Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical | ChemComp-PGO / | 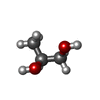  Mass: 76.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O2 Mass: 76.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 165 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 165 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.88 Å3/Da / Density % sol: 57.34 % / Description: Prismatic crystals |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7.6 Details: Protein: redissolved from lyophilized at 5mg/ml mamb-1-T23A in 50mM Na Acetate, pH 5.5. Precipitant: 21.6% PEG600, 3.6% PEG 20K 0.18M mixed L-malic acid, MES, Tris (pH4) and mixed sodium ...Details: Protein: redissolved from lyophilized at 5mg/ml mamb-1-T23A in 50mM Na Acetate, pH 5.5. Precipitant: 21.6% PEG600, 3.6% PEG 20K 0.18M mixed L-malic acid, MES, Tris (pH4) and mixed sodium Malonate, imidazole, boric acid(pH10) in the ratio 40:60. Cryoprotectant: 30% MPEG 550, 40% CryoProtX cryomix9, 10% NaAc pH 6.5, 0.1M potassium iodide soaked for 1 min. PH range: 5.5 - 7.6 / Temp details: cooled incubator |
-Data collection
| Diffraction | Mean temperature: 100 K / Ambient temp details: N2 cryostat - Helical scan |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL  / Beamline: PROXIMA 2 / Wavelength: 0.9801 Å / Beamline: PROXIMA 2 / Wavelength: 0.9801 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Jun 3, 2015 / Details: X-ray centering - Helical scan |
| Radiation | Monochromator: [111] Si Cut monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9801 Å / Relative weight: 1 |
| Reflection | Resolution: 1.697→37 Å / Num. all: 16719 / Num. obs: 31543 / % possible obs: 97.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 7.1 % / Rmerge(I) obs: 0.061 / Rsym value: 0.057 / Net I/σ(I): 21.83 |
| Reflection shell | Resolution: 1.697→1.74 Å / Redundancy: 4.8 % / Rmerge(I) obs: 0.408 / Mean I/σ(I) obs: 4.64 / % possible all: 78.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 1.697→27.95 Å / SU ML: 0.16 / Cross valid method: THROUGHOUT / σ(F): 1.38 / Phase error: 21.25 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.697→27.95 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
