 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5dk6: CRYSTAL STRUCTURE OF A 5'-METHYLTHIOADENOSINE/S-ADENOSYLHOMOCYSTE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5dk6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A 5'-METHYLTHIOADENOSINE/S-ADENOSYLHOMOCYSTEINE (MTA/SAH) NUCLEOSIDASE (MTAN) FROM COLWELLIA PSYCHRERYTHRAEA 34H (CPS_4743, TARGET PSI-029300) IN COMPLEX WITH ADENINE AT 2.27 A RESOLUTION | ||||||
 Components Components | 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase | ||||||
 Keywords Keywords | HYDROLASE / MTA/SAH NUCLEOSIDASE FAMILY / MTAN / GAMMAPROTEOBACTERIA / Vibrio psychroerythus / ADENINE / PROTEIN-LIGAND COMPLEX / PROTEIN STRUCTURE INITIATIVE / PSI-Biology / Structural Genomics / New York Structural Genomics Research Consortium / NYSGRC | ||||||
| Function / homology |  Function and homology information Function and homology informationadenosylhomocysteine nucleosidase / adenosylhomocysteine nucleosidase activity / methylthioadenosine nucleosidase activity / : / nucleoside catabolic process / : / cytosol Similarity search - Function | ||||||
| Biological species |  Colwellia psychrerythraea (bacteria) Colwellia psychrerythraea (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.27 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.27 Å | ||||||
 Authors Authors | Himmel, D.M. / Bhosle, R. / Toro, R. / Ahmed, M. / Hillerich, B. / Gizzi, A. / Garforth, S. / Kar, A. / Chan, M.K. / Lafluer, J. ...Himmel, D.M. / Bhosle, R. / Toro, R. / Ahmed, M. / Hillerich, B. / Gizzi, A. / Garforth, S. / Kar, A. / Chan, M.K. / Lafluer, J. / Patel, H. / Matikainen, B. / Chamala, S. / Lim, S. / Celikgil, A. / Villegas, G. / Evans, B. / Love, J. / Fiser, A. / Seidel, R.D. / Bonanno, J.B. / Almo, S.C. / New York Structural Genomics Research Consortium (NYSGRC) | ||||||
 Citation Citation | Journal: To be published Title: CRYSTAL STRUCTURE OF A 5'-METHYLTHIOADENOSINE/S-ADENOSYLHOMOCYSTEINE (MTA/SAH)NUCLEOSIDASE (MTAN) FROM COLWELLIA PSYCHRERYTHRAEA 34H (CPS_4743, TARGET PSI-029300) IN COMPLEX WITH ADENINE AT 2.27 A RESOLUTION Authors: Himmel, D.M. / Bhosle, R. / Toro, R. / Ahmed, M. / Hillerich, B. / Gizzi, A. / Garforth, S. / Kar, A. / Chan, M.K. / Lafluer, J. / Patel, H. / Matikainen, B. / Chamala, S. / Lim, S. / ...Authors: Himmel, D.M. / Bhosle, R. / Toro, R. / Ahmed, M. / Hillerich, B. / Gizzi, A. / Garforth, S. / Kar, A. / Chan, M.K. / Lafluer, J. / Patel, H. / Matikainen, B. / Chamala, S. / Lim, S. / Celikgil, A. / Villegas, G. / Evans, B. / Love, J. / Fiser, A. / Seidel, R.D. / Bonanno, J.B. / Almo, S.C. / New York Structural Genomics Research Consortium (NYSGRC) #1: Journal: Biochemistry / Year: 2015Title: Active site and remote contributions to catalysis in methylthioadenosine nucleosidases. Authors: Thomas, K. / Cameron, S.A. / Almo, S.C. / Burgos, E.S. / Gulab, S.A. / Schramm, V.L. #2: Journal: Nat. Chem. Biol. / Year: 2009Title: Transition state analogs of 5'-methylthioadenosine nucleosidase disrupt quorum sensing. Authors: Gutierrez, J.A. / Crowder, T. / Rinaldo-Matthis, A. / Ho, M.C. / Almo, S.C. / Schramm, V.L. | ||||||
| History |
|
- Structure visualization
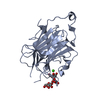
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5dk6.cif.gz | 63.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5dk6.ent.gz | 45.8 KB | Display | PDB format |
| PDBx/mmJSON format | 5dk6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dk/5dk6ftp://data.pdbj.org/pub/pdb/validation_reports/dk/5dk6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data | |
| Other databases |














-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| 2 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 28485.967 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Colwellia psychrerythraea (bacteria) / Strain: 34H / ATCC BAA-681 / Gene: mtnN, CPS_4743 / Plasmid: pET / Production host: References: UniProt: Q47UY5, adenosylhomocysteine nucleosidase |
|---|---|
| #2: Chemical | ChemComp-GLY / 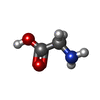  Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H5NO2 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H5NO2 |
| #3: Chemical | ChemComp-ADE /   Mass: 135.127 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: C5H5N5 Mass: 135.127 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: C5H5N5 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 101 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 101 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.84 Å3/Da / Density % sol: 68 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, sitting drop / pH: 8 Details: Protein (37.95 mg/ml, 20 mM HEPES pH 7.5, 150 mM Sodium Chloride, 5% v/v Glycerol, 5 mM DTT) were combined with an equal volume of Reservoir (100 mM Bicine pH 8.0, 1.98 M Ammonium Sulfate, ...Details: Protein (37.95 mg/ml, 20 mM HEPES pH 7.5, 150 mM Sodium Chloride, 5% v/v Glycerol, 5 mM DTT) were combined with an equal volume of Reservoir (100 mM Bicine pH 8.0, 1.98 M Ammonium Sulfate, 135 mM Sodium Succinate, 4 mM Glycine, 15 mM CYMAL-7); Cryoprotection (75 mM Bicine pH 8.0, 1.49 M Ammonium Sulfate, 101 mM Sodium Succinate, 3 mM Glycine, 11 mM CYMAL-7) |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 31-ID / Wavelength: 0.97931 Å / Beamline: 31-ID / Wavelength: 0.97931 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RAYONIX MX225HE / Detector: CCD / Date: Oct 17, 2013 / Details: MIRRORS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97931 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 3.9 % / Number: 143135 / Rmerge(I) obs: 0.1 / Χ2: 1 / D res high: 2.28 Å / D res low: 26 Å / Num. obs: 37023 / % possible obs: 96.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.268→26 Å / Num. obs: 37023 / % possible obs: 96.4 % / Observed criterion σ(I): -0.3 / Redundancy: 3.9 % / Biso Wilson estimate: 33.16 Å2 / Rmerge(I) obs: 0.1 / Net I/σ(I): 11.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 2.27→2.36 Å / Redundancy: 2 % / Rmerge(I) obs: 0.337 / Mean I/σ(I) obs: 2.148 / % possible all: 82.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.27→25.28 Å / SU ML: 0.23 / Cross valid method: FREE R-VALUE / σ(F): 1.45 / Phase error: 22.27 / Stereochemistry target values: MLHL
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 31.6 Å2 / ksol: 0.34 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 43.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.27→25.28 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
