 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5a9w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of GDPCP BipA | ||||||
 Components Components | GTP-BINDING PROTEIN | ||||||
 Keywords Keywords | RIBOSOMAL PROTEIN / RIBOSOME / TRANSLATIONAL GTPASE FACTORS | ||||||
| Function / homology |  Function and homology information Function and homology informationguanosine tetraphosphate binding / protein folding chaperone / response to cold / response to heat / ribosome binding / gene expression / ribosomal large subunit assembly / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / tRNA binding / rRNA binding ...guanosine tetraphosphate binding / protein folding chaperone / response to cold / response to heat / ribosome binding / gene expression / ribosomal large subunit assembly / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / tRNA binding / rRNA binding / translation / ribonucleoprotein complex / GTPase activity / GTP binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.7 Å | ||||||
 Authors Authors | Kumar, V. / Chen, Y. / Ero, R. / Li, Z. / Gao, Y. | ||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2015 Title: Structure of BipA in GTP form bound to the ratcheted ribosome. Authors: Veerendra Kumar / Yun Chen / Rya Ero / Tofayel Ahmed / Jackie Tan / Zhe Li / Andrew See Weng Wong / Shashi Bhushan / Yong-Gui Gao /  Abstract: BPI-inducible protein A (BipA) is a member of the family of ribosome-dependent translational GTPase (trGTPase) factors along with elongation factors G and 4 (EF-G and EF4). Despite being highly ...BPI-inducible protein A (BipA) is a member of the family of ribosome-dependent translational GTPase (trGTPase) factors along with elongation factors G and 4 (EF-G and EF4). Despite being highly conserved in bacteria and playing a critical role in coordinating cellular responses to environmental changes, its structures (isolated and ribosome bound) remain elusive. Here, we present the crystal structures of apo form and GTP analog, GDP, and guanosine-3',5'-bisdiphosphate (ppGpp)-bound BipA. In addition to having a distinctive domain arrangement, the C-terminal domain of BipA has a unique fold. Furthermore, we report the cryo-electron microscopy structure of BipA bound to the ribosome in its active GTP form and elucidate the unique structural attributes of BipA interactions with the ribosome and A-site tRNA in the light of its possible function in regulating translation. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5a9w.cif.gz | 123.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5a9w.ent.gz | 96.7 KB | Display | PDB format |
| PDBx/mmJSON format | 5a9w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a9/5a9wftp://data.pdbj.org/pub/pdb/validation_reports/a9/5a9w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6396C  6397C  5a9vC  5a9xC  5a9yC  5a9zC  5aa0C  3e3xS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 67439.312 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-GCP / 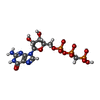  Mass: 521.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O13P3 / Comment: GMP-PCP, energy-carrying molecule analogue*YM Mass: 521.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O13P3 / Comment: GMP-PCP, energy-carrying molecule analogue*YM |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.1 Å3/Da / Density % sol: 70 % / Description: NONE |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 / Beamline: X06SA / Wavelength: 1 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jul 17, 2014 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.66→48.49 Å / Num. obs: 12870 / % possible obs: 99.5 % / Observed criterion σ(I): 2.1 / Redundancy: 18.7 % / Rmerge(I) obs: 0.14 / Net I/σ(I): 14.3 |
| Reflection shell | Resolution: 3.66→3.86 Å / Redundancy: 17.3 % / Rmerge(I) obs: 1.45 / Mean I/σ(I) obs: 2.1 / % possible all: 97.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3E3X Resolution: 3.7→48.49 Å / Cor.coef. Fo:Fc: 0.93 / Cor.coef. Fo:Fc free: 0.887 / SU B: 47.304 / SU ML: 0.66 / Cross valid method: THROUGHOUT / ESU R Free: 0.718 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 152.456 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.7→48.49 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|