 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4wc1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of tRNA-processing enzyme with CTP | ||||||
 Components Components | Poly A polymerase | ||||||
 Keywords Keywords | TRANSFERASE / RNA Nucleotidyltransferase / CCA-adding enzyme / A-adding enzyme | ||||||
| Function / homology |  Function and homology information Function and homology informationATP:3'-cytidine-cytidine-tRNA adenylyltransferase activity / tRNA processing / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / tRNA binding / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |   Aquifex aeolicus (bacteria) Aquifex aeolicus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3.1 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3.1 Å | ||||||
 Authors Authors | Yamashita, S. / Tomita, K. | ||||||
| Funding support |  Japan, 1items Japan, 1items
| ||||||
 Citation Citation | Journal: Structure / Year: 2015 Title: Measurement of Acceptor-T Psi C Helix Length of tRNA for Terminal A76-Addition by A-Adding Enzyme. Authors: Yamashita, S. / Martinez, A. / Tomita, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4wc1.cif.gz | 321.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4wc1.ent.gz | 262.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4wc1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wc/4wc1ftp://data.pdbj.org/pub/pdb/validation_reports/wc/4wc1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4wbySC  4wbzC  4wc0C  4wc2C  4wc3C  4wc4C  4wc5C  4wc6C  4wc7C  4x0aC  4x0bC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 46550.711 Da / Num. of mol.: 2 / Fragment: UNP RESIDUES 443-824 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Aquifex aeolicus (bacteria) / Strain: VF5 / Gene: pcnB1, aq_411 / Plasmid: pET22b / Production host: #2: Chemical | 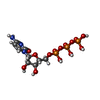  Mass: 483.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H16N3O14P3 Mass: 483.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H16N3O14P3 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.7 Å3/Da / Density % sol: 54.51 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / Details: PEGMME550, sodium chloride, Tris-Cl |
-Data collection
| Diffraction | Mean temperature: 95 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory / Beamline: BL-17A / Wavelength: 0.97897 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 270 / Detector: CCD / Date: Jan 25, 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97897 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection twin | Operator: h,-k,-h-l / Fraction: 0.49 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 3.1→50 Å / Num. obs: 17858 / % possible obs: 98.2 % / Observed criterion σ(I): -3 / Redundancy: 3.8 % / Rmerge F obs: 0.998 / Rmerge(I) obs: 0.076 / Rrim(I) all: 0.088 / Χ2: 0.938 / Net I/σ(I): 16.08 / Num. measured all: 67902 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Starting model: 4WBY Resolution: 3.1→19.833 Å / FOM work R set: 0.6607 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 24.88 / Stereochemistry target values: TWIN_LSQ_F
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 438.49 Å2 / Biso mean: 135.59 Å2 / Biso min: 49.63 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 3.1→19.833 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 6
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
