 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4utn: Crystal structure of zebrafish Sirtuin 5 in complex with succinyl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4utn | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of zebrafish Sirtuin 5 in complex with succinylated CPS1-peptide | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / REGULATORY ENZYME / ROSSMANN-FOLD / ZINC-BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationTranscriptional activation of mitochondrial biogenesis / regulation of ketone biosynthetic process / peptidyl-lysine demalonylation / protein desuccinylation / peptidyl-lysine desuccinylation / protein-glutaryllysine deglutarylase activity / protein-malonyllysine demalonylase activity / protein-succinyllysine desuccinylase activity / histone deacetylase activity, NAD-dependent / heterocyclic compound binding ...Transcriptional activation of mitochondrial biogenesis / regulation of ketone biosynthetic process / peptidyl-lysine demalonylation / protein desuccinylation / peptidyl-lysine desuccinylation / protein-glutaryllysine deglutarylase activity / protein-malonyllysine demalonylase activity / protein-succinyllysine desuccinylase activity / histone deacetylase activity, NAD-dependent / heterocyclic compound binding / acyl binding / NAD+ binding / Transferases; Acyltransferases; Transferring groups other than aminoacyl groups / mitochondrial matrix / mitochondrion / zinc ion binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |   HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3 Å | ||||||
 Authors Authors | Pannek, M. / Gertz, M. / Steegborn, C. | ||||||
 Citation Citation | Journal: Angew.Chem.Int.Ed.Engl. / Year: 2014 Title: Chemical Probing of the Human Sirtuin 5 Active Site Reveals its Substrate Acyl Specificity and Peptide-Based Inhibitors. Authors: Roessler, C. / Nowak, T. / Pannek, M. / Gertz, M. / Nguyen, G.T. / Scharfe, M. / Born, I. / Sippl, W. / Steegborn, C. / Schutkowski, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4utn.cif.gz | 222.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4utn.ent.gz | 180.1 KB | Display | PDB format |
| PDBx/mmJSON format | 4utn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ut/4utnftp://data.pdbj.org/pub/pdb/validation_reports/ut/4utn | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 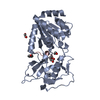 4utrC  4utvC  4utxC  4utzC  4uu7C  4uu8C  4uuaC  4uubC  2nyrS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.74826, -0.5507, -0.36991), Vector: |
-Components
-Protein / Protein/peptide , 2 types, 3 molecules ABD
| #1: Protein | Mass: 30423.785 Da / Num. of mol.: 2 / Fragment: CATALYTIC CORE, RESIDUES 30-298 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: Q6DHI5, Hydrolases; Acting on carbon-nitrogen bonds, other than peptide bonds; In linear amides #2: Protein/peptide | | Mass: 1068.177 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) HOMO SAPIENS (human) |
|---|
-Non-polymers , 5 types, 22 molecules 




| #3: Chemical |  Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-EPE / |  Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#5: Chemical | ChemComp-DMS / |  Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#6: Chemical | ChemComp-EDO / |  Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 17 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | BENZOYLATE |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.86 Å3/Da / Density % sol: 57 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.4 / Details: 20% PEG3350, 0.1 M HEPES PH 7.4 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.9184 / Beamline: 14.1 / Wavelength: 0.9184 |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Nov 14, 2012 / Details: MIRROR |
| Radiation | Monochromator: SI111 CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 3→100 Å / Num. obs: 15155 / % possible obs: 99.9 % / Observed criterion σ(I): 1.7 / Redundancy: 8.1 % / Rmerge(I) obs: 0.18 / Net I/σ(I): 11.2 |
| Reflection shell | Resolution: 3→3.1 Å / Redundancy: 8.5 % / Rmerge(I) obs: 1.34 / Mean I/σ(I) obs: 1.7 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2NYR Resolution: 3→75.73 Å / Cor.coef. Fo:Fc: 0.924 / Cor.coef. Fo:Fc free: 0.902 / SU B: 43.64 / SU ML: 0.381 / Cross valid method: THROUGHOUT / ESU R Free: 0.44 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES WITH TLS ADDED. STRUCTURE REFINED USING PROSMART WITH PDB-ID 3RIY R.A. NICHOLLS, F. LONG AND G.N. MURSHUDOV (2012) LOW RESOLUTION ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES WITH TLS ADDED. STRUCTURE REFINED USING PROSMART WITH PDB-ID 3RIY R.A. NICHOLLS, F. LONG AND G.N. MURSHUDOV (2012) LOW RESOLUTION REFINEMENT TOOLS IN REFMAC5. ACTA CRYST. D.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 70.934 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→75.73 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|