 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4tpi: THE REFINED 2.2-ANGSTROMS (0.22-NM) X-RAY CRYSTAL STRUCTURE OF TH... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4tpi | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE REFINED 2.2-ANGSTROMS (0.22-NM) X-RAY CRYSTAL STRUCTURE OF THE TERNARY COMPLEX FORMED BY BOVINE TRYPSINOGEN, VALINE-VALINE AND THE ARG15 ANALOGUE OF BOVINE PANCREATIC TRYPSIN INHIBITOR | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / COMPLEX (PROTEINASE-INHIBITOR) / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationsulfate binding / negative regulation of platelet aggregation / zymogen binding / potassium channel inhibitor activity / molecular function inhibitor activity / negative regulation of thrombin-activated receptor signaling pathway / trypsin / serpin family protein binding / serine protease inhibitor complex / digestion ...sulfate binding / negative regulation of platelet aggregation / zymogen binding / potassium channel inhibitor activity / molecular function inhibitor activity / negative regulation of thrombin-activated receptor signaling pathway / trypsin / serpin family protein binding / serine protease inhibitor complex / digestion / serine-type endopeptidase inhibitor activity / protease binding / endopeptidase activity / serine-type endopeptidase activity / calcium ion binding / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.2 Å X-RAY DIFFRACTION / Resolution: 2.2 Å | ||||||
 Authors Authors | Bode, W. / Walter, J. | ||||||
 Citation Citation | Journal: Eur.J.Biochem. / Year: 1984 Title: The refined 2.2-A (0.22-nm) X-ray crystal structure of the ternary complex formed by bovine trypsinogen, valine-valine and the Arg15 analogue of bovine pancreatic trypsin inhibitor Authors: Bode, W. / Walter, J. / Huber, R. / Wenzel, H.R. / Tschesche, H. #1: Journal: J.Mol.Biol. / Year: 1979Title: The Transition of Bovine Trypsinogen to a Trypsin-Like State Upon Strong Ligand Binding. II. The Binding of the Pancreatic Trypsin Inhibitor and of Isoleucine-Valine and of Sequentially ...Title: The Transition of Bovine Trypsinogen to a Trypsin-Like State Upon Strong Ligand Binding. II. The Binding of the Pancreatic Trypsin Inhibitor and of Isoleucine-Valine and of Sequentially Related Peptides to Trypsinogen and to P-Guanidinobenzoate-Trypsinogen Authors: Bode, W. #2: Journal: J.Mol.Biol. / Year: 1978Title: The Transition of Bovine Trypsinogen to a Trypsin-Like State Upon Strong Ligand Binding. The Refined Crystal Structures of the Bovine Trypsinogen-Pancreatic Trypsin Inhibitor Complex and of ...Title: The Transition of Bovine Trypsinogen to a Trypsin-Like State Upon Strong Ligand Binding. The Refined Crystal Structures of the Bovine Trypsinogen-Pancreatic Trypsin Inhibitor Complex and of its Ternary Complex with Ile-Val at 1.9 Angstroms Resolution Authors: Bode, W. / Schwager, P. / Huber, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4tpi.cif.gz | 68.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4tpi.ent.gz | 50.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4tpi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tp/4tpiftp://data.pdbj.org/pub/pdb/validation_reports/tp/4tpi | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: SEE REMARK 4. / 2: SEE REMARK 7. / 3: SEE REMARK 8. |
-Components
-Protein , 2 types, 2 molecules ZI
| #1: Protein | Mass: 24012.953 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Protein | Mass: 6555.582 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source References: UniProt: P00974 |
-Non-polymers , 4 types, 160 molecules 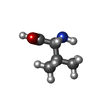



| #3: Chemical |  Type: L-peptide linking / Mass: 117.146 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H11NO2 Type: L-peptide linking / Mass: 117.146 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H11NO2#4: Chemical | ChemComp-CA / |  Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#5: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 155 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Compound details | RESIDUE SER Z 195 HAS BEEN ALLOWED TO ACCESS INHIBITOR RESIDUES FREELY, I. E. IT IS NOT RESTRICTED ...RESIDUE SER Z 195 HAS BEEN ALLOWED TO ACCESS INHIBITOR RESIDUES FREELY, I. E. IT IS NOT RESTRICTED |
|---|---|
| Has protein modification | Y |
| Nonpolymer details | THE RESIDUES 1016 AND 1017 REPRESENT A DIPEPTIDE (VAL-VAL) BOUND TO THE ENZYME THE 229 AMINO ACIDS ...THE RESIDUES 1016 AND 1017 REPRESENT A DIPEPTIDE (VAL-VAL) BOUND TO THE ENZYME THE 229 AMINO ACIDS OF TRYPSINOGE |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.2 Å3/Da / Density % sol: 61.6 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 6.9 / Method: vapor diffusion | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.24 Å / Num. obs: 12422 / % possible obs: 63.8 % / Observed criterion σ(I): 1 / Num. measured all: 18470 / Rmerge(I) obs: 0.069 |
- Processing
Processing
| Software | Name: EREF / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.2→7 Å / Rfactor Rwork: 0.17 Details: THE SIDE CHAIN CONFORMATION OF VAL 1016 AS DESCRIBED IN THE JRNL REFERENCE ABOVE AND AS GIVEN ON THE ATOMS RECORDS BELOW IS ENERGETICALLY UNFAVORABLE. A MORE FAVORABLE CONFORMATION COULD BE ...Details: THE SIDE CHAIN CONFORMATION OF VAL 1016 AS DESCRIBED IN THE JRNL REFERENCE ABOVE AND AS GIVEN ON THE ATOMS RECORDS BELOW IS ENERGETICALLY UNFAVORABLE. A MORE FAVORABLE CONFORMATION COULD BE OBTAINED BY A 180 DEGREE SIDE CHAIN ROTATION ABOUT CHI 1 (VAL 1016 CA - CB). THE SIDE CHAIN CONFORMATION OF VAL S 16 AS DESCRIBED IN THE JRNL REFERENCE ABOVE AND AS GIVEN ON THE ATOMS RECORDS BELOW IS ENERGETICALLY UNFAVORABLE. A MORE FAVORABLE CONFORMATION COULD BE OBTAINED BY A 180 DEGREE SIDE CHAIN ROTATION ABOUT CHI 1 (VAL S 16 CA - CB). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.24 Å / Rfactor Rwork: 0.17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
