 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4pth: Ensemble model for Escherichia coli dihydrofolate reductase at 100K -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4pth | ||||||
|---|---|---|---|---|---|---|---|
| Title | Ensemble model for Escherichia coli dihydrofolate reductase at 100K | ||||||
 Components Components | Dihydrofolate reductase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / ROSSMANN FOLD | ||||||
| Function / homology | Dihydrofolate Reductase, subunit A / Dihydrofolate Reductase, subunit A / 3-Layer(aba) Sandwich / Alpha Beta / FOLIC ACID / : / NADP NICOTINAMIDE-ADENINE-DINUCLEOTIDE PHOSPHATE / :  Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.85 Å | ||||||
 Authors Authors | Keedy, D.A. / van den Bedem, H. / Sivak, D.A. / Petsko, G.A. / Ringe, D. / Wilson, M.A. / Fraser, J.S. | ||||||
 Citation Citation | Journal: Structure / Year: 2014 Title: Crystal Cryocooling Distorts Conformational Heterogeneity in a Model Michaelis Complex of DHFR. Authors: Keedy, D.A. / van den Bedem, H. / Sivak, D.A. / Petsko, G.A. / Ringe, D. / Wilson, M.A. / Fraser, J.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4pth.cif.gz | 28.8 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4pth.ent.gz | 24.5 MB | Display | PDB format |
| PDBx/mmJSON format | 4pth.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pt/4pthftp://data.pdbj.org/pub/pdb/validation_reports/pt/4pth | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4p3qC  4p3rC  4pssC  4pstC  4ptjC  1rx2S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Number of models | 250 |
-Components
| #1: Protein | Mass: 18051.338 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-FOL / 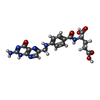  Mass: 441.397 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H19N7O6 Mass: 441.397 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H19N7O6 | ||||
| #3: Chemical | ChemComp-NAP /   Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 | ||||
| #4: Chemical |   Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.07 Å3/Da / Density % sol: 40.62 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 17% PEG 400, 20 mM imidazole pH 7.0, 125 mM MnCl2, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL11-1 / Wavelength: 0.9 Å / Beamline: BL11-1 / Wavelength: 0.9 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Apr 25, 2005 |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 0.85→49 Å / Num. all: 130200 / Num. obs: 130200 / % possible obs: 98.2 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 5.5 % / Biso Wilson estimate: 7.7 Å2 / Rmerge(I) obs: 0.049 / Net I/σ(I): 36.3 |
| Reflection shell | Resolution: 0.85→0.88 Å / Redundancy: 4 % / Rmerge(I) obs: 0.55 / Mean I/σ(I) obs: 2.3 / Num. unique all: 12715 / % possible all: 97.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1RX2 Resolution: 0.85→49 Å / σ(F): 0 / Stereochemistry target values: Engh & Huber / Details: Ensemble refinement as implemented in PHENIX
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.85→49 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 0.8495→0.8591 Å
|
