 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4psb: Crystal Structure of Phytohormone Binding Protein from Vigna radi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4psb | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Phytohormone Binding Protein from Vigna radiata in complex with gibberellic acid (GA3) | ||||||
 Components Components | Cytokinin-specific binding protein | ||||||
 Keywords Keywords | PLANT PROTEIN / Cytokinin Specific Binding Protein (CSBP) / PR-10 FOLD / GIBBERELLIN / CYTOKININ / PLANT HORMONE BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationcytokinin binding / gibberellin binding / abscisic acid binding / abscisic acid-activated signaling pathway / protein phosphatase inhibitor activity / defense response / signaling receptor activity / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  Vigna radiata (mung bean) Vigna radiata (mung bean) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.42 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.42 Å | ||||||
 Authors Authors | Ruszkowski, M. / Sikorski, M. / Jaskolski, M. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2014 Title: Specific binding of gibberellic acid by Cytokinin-Specific Binding Proteins: a new aspect of plant hormone-binding proteins with the PR-10 fold. Authors: Ruszkowski, M. / Sliwiak, J. / Ciesielska, A. / Barciszewski, J. / Sikorski, M. / Jaskolski, M. #1: Journal: Plant Cell / Year: 2006Title: Crystal structure of Vigna radiata cytokinin-specific binding protein in complex with zeatin. Authors: Pasternak, O. / Bujacz, G.D. / Fujimoto, Y. / Hashimoto, Y. / Jelen, F. / Otlewski, J. / Sikorski, M.M. / Jaskolski, M. #2: Journal: J.Mol.Biol. / Year: 2002Title: Crystal structures of two homologous pathogenesis-related proteins from yellow lupine. Authors: Biesiadka, J. / Bujacz, G. / Sikorski, M.M. / Jaskolski, M. #3: Journal: Acta Crystallogr.,Sect.D / Year: 2013Title: The landscape of cytokinin binding by a plant nodulin. Authors: Ruszkowski, M. / Szpotkowski, K. / Sikorski, M. / Jaskolski, M. #4: Journal: J.Mol.Biol. / Year: 2008Title: Lupinus luteus pathogenesis-related protein as a reservoir for cytokinin. Authors: Fernandes, H. / Pasternak, O. / Bujacz, G. / Bujacz, A. / Sikorski, M.M. / Jaskolski, M. #5: Journal: Febs J. / Year: 2013 Title: Structural and functional aspects of PR-10 proteins. Authors: Fernandes, H. / Michalska, K. / Sikorski, M. / Jaskolski, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4psb.cif.gz | 85.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4psb.ent.gz | 63.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4psb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ps/4psbftp://data.pdbj.org/pub/pdb/validation_reports/ps/4psb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4q0kC  2flhS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 17612.852 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Vigna radiata (mung bean) / Gene: vrphbp / Plasmid: PET 3A / Production host:  |
|---|---|
| #2: Chemical | ChemComp-GA3 / 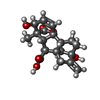  Mass: 346.374 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H22O6 Mass: 346.374 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H22O6 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 140 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 140 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.86 Å3/Da / Density % sol: 33.93 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 4 Details: 0.1 M MMT buffer pH 4.0, 25% PEG 1500, VAPOR DIFFUSION, HANGING DROP, temperature 292K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY  / Beamline: P14 (MX2) / Wavelength: 0.97551 Å / Beamline: P14 (MX2) / Wavelength: 0.97551 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Sep 12, 2013 / Details: mirrors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Double crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97551 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.42→35.14 Å / Num. all: 24476 / Num. obs: 24350 / % possible obs: 99.5 % / Observed criterion σ(I): -3 / Redundancy: 4.9 % / Biso Wilson estimate: 18.06 Å2 / Rmerge(I) obs: 0.079 / Net I/σ(I): 9.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2FLH Resolution: 1.42→35.14 Å / SU ML: 0.18 / Phase error: 25.01 / Stereochemistry target values: ENGH & HUBER Details: HYDROGEN ATOMS WERE ADDED AT RIDING POSITIONS. ANISOTROPIC REFINEMENT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.53 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.42→35.14 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
