| Entry | Database: PDB / ID: 4pq0
|
|---|
| Title | Crystal structure of POB3 middle domain at 1.65A |
|---|
 Components Components | FACT complex subunit POB3 |
|---|
 Keywords Keywords | REPLICATION / TRANSCRIPTION / double pleckstrin homology (PH)architecture |
|---|
| Function / homology |  Function and homology information Function and homology information
Regulation of TP53 Activity through Phosphorylation / FACT complex / regulation of chromatin organization / nucleosome organization / TP53 Regulates Transcription of DNA Repair Genes / RNA Polymerase II Pre-transcription Events / positive regulation of RNA polymerase II transcription preinitiation complex assembly / DNA-templated DNA replication / chromatin organization / DNA repair ...Regulation of TP53 Activity through Phosphorylation / FACT complex / regulation of chromatin organization / nucleosome organization / TP53 Regulates Transcription of DNA Repair Genes / RNA Polymerase II Pre-transcription Events / positive regulation of RNA polymerase II transcription preinitiation complex assembly / DNA-templated DNA replication / chromatin organization / DNA repair / chromatin / DNA bindingSimilarity search - Function PH-domain like - #150 / : / SSRP1 PH domain / FACT complex subunit SSRP1/POB3 / SSRP1, dimerization domain / FACT complex subunit SSRP1/POB3, N-terminal PH domain / SSRP1 domain superfamily / : / Structure-specific recognition protein (SSRP1) / POB3-like N-terminal PH domain ...PH-domain like - #150 / : / SSRP1 PH domain / FACT complex subunit SSRP1/POB3 / SSRP1, dimerization domain / FACT complex subunit SSRP1/POB3, N-terminal PH domain / SSRP1 domain superfamily / : / Structure-specific recognition protein (SSRP1) / POB3-like N-terminal PH domain / Histone chaperone RTT106/FACT complex subunit SPT16-like, middle domain / Histone chaperone Rttp106-like, middle domain / Histone chaperone Rttp106-like / Pleckstrin-homology domain (PH domain)/Phosphotyrosine-binding domain (PTB) / PH-domain like / PH-like domain superfamily / Roll / Mainly BetaSimilarity search - Domain/homology |
|---|
| Biological species |   Saccharomyces cerevisiae (brewer's yeast) Saccharomyces cerevisiae (brewer's yeast) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å |
|---|
 Authors Authors | Liu, H. |
|---|
 Citation Citation | Journal: TO BE PUBLISHED
Title: Crystal structure of POB3 middle domain at 1.65A
Authors: Liu, H. |
|---|
| History | | Deposition | Feb 28, 2014 | Deposition site: RCSB / Processing site: PDBJ |
|---|
| Revision 1.0 | Mar 26, 2014 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Apr 29, 2015 | Group: Non-polymer description |
|---|
| Revision 1.2 | Mar 20, 2024 | Group: Data collection / Database references / Derived calculations
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj




 Assembly
Assembly

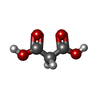
 Mass: 104.061 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H4O4
Mass: 104.061 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H4O4 Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL17U
/ Beamline: BL17U Processing
Processing