 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
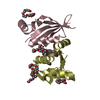
| Entry | Database: PDB / ID: 4nxf | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of iLOV-I486(2LT) at pH 8.0 | ||||||
 Components Components | Phototropin-2 | ||||||
 Keywords Keywords | FLAVOPROTEIN / FLUORESCENT PROTEIN / FMN binding | ||||||
| Function / homology |  Function and homology information Function and homology informationchloroplast relocation / phototropism / blue light signaling pathway / stomatal movement / blue light photoreceptor activity / response to blue light / plastid / circadian rhythm / kinase activity / FMN binding ...chloroplast relocation / phototropism / blue light signaling pathway / stomatal movement / blue light photoreceptor activity / response to blue light / plastid / circadian rhythm / kinase activity / FMN binding / non-specific serine/threonine protein kinase / protein serine kinase activity / protein serine/threonine kinase activity / Golgi apparatus / ATP binding / membrane / identical protein binding / nucleus / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.766 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.766 Å | ||||||
 Authors Authors | Wang, J. / Liu, X. / Li, J. | ||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2014 Title: Significant expansion of fluorescent protein sensing ability through the genetic incorporation of superior photo-induced electron-transfer quenchers. Authors: Liu, X. / Jiang, L. / Li, J. / Wang, L. / Yu, Y. / Zhou, Q. / Lv, X. / Gong, W. / Lu, Y. / Wang, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4nxf.cif.gz | 109 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4nxf.ent.gz | 82.1 KB | Display | PDB format |
| PDBx/mmJSON format | 4nxf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nx/4nxfftp://data.pdbj.org/pub/pdb/validation_reports/nx/4nxf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4nx2C  4nxbC  4nxeC  4nxgC  4eesS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13969.408 Da / Num. of mol.: 2 / Fragment: LOV DOMAIN, UNP Residues 388-496 / Mutation: S394T, S409G, I452T, F470L, M475V, I486Y Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: P93025, non-specific serine/threonine protein kinase #2: Chemical | 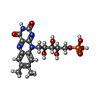  Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 108 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 108 / Source method: isolated from a natural source / Formula: H2OSequence details | RESIDUES C426A IS MUTAGENESI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.08 Å3/Da / Density % sol: 40.96 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, sitting drop / pH: 8 Details: protein sample (20-30 mg/ml) in 20mM Tris, pH 8.0, 50mM NaCl, equal volume of reservoir solution (0.2M Ammonium acetate, 0.1M Tris pH 8.0, 16% w/v Polyethylene glycol 10000), VAPOR ...Details: protein sample (20-30 mg/ml) in 20mM Tris, pH 8.0, 50mM NaCl, equal volume of reservoir solution (0.2M Ammonium acetate, 0.1M Tris pH 8.0, 16% w/v Polyethylene glycol 10000), VAPOR DIFFUSION, SITTING DROP, temperature 289.0 K |
-Data collection
| Diffraction | Mean temperature: 200 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF  / Beamline: BL17U / Wavelength: 0.979 Å / Beamline: BL17U / Wavelength: 0.979 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Jan 20, 2013 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 1.766→50 Å / Num. all: 22542 / Num. obs: 21167 / % possible obs: 93.9 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 |
| Reflection shell | Resolution: 1.766→1.8 Å / Redundancy: 3.6 % / Num. unique all: 1066 / % possible all: 98.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4EES Resolution: 1.766→24.203 Å / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.8606 / SU ML: 0.16 / σ(F): 1.43 / Phase error: 21.58 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 70.74 Å2 / Biso mean: 24.1841 Å2 / Biso min: 11.02 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.766→24.203 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -3.5914 Å / Origin y: -2.4953 Å / Origin z: 60.3517 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: ALL |
