 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4nhc: Crystal structure of the HIV-1 neutralizing antibody 4E10 Fab fra... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4nhc | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the HIV-1 neutralizing antibody 4E10 Fab fragment in complex with a hydrocarbon-stapled peptide containing the 4e10 epitope on gp41. | ||||||
 Components Components |
| ||||||
 Keywords Keywords | IMMUNE SYSTEM / IMMUNOGLOBULIN FOLD / BETA-SANDWICH / 4E10 FAB / ANTIBODY-EPITOPE COMPLEX / GP41 HIV-1 / HYDROCARBON STAPLE | ||||||
| Function / homology | Immunoglobulins / Immunoglobulin-like / Sandwich / Mainly Beta / Stapled peptide (ACE)NWFNITN(DIV)LW(MK8)IKKKK / PHOSPHATE ION / trifluoroacetic acid Function and homology information Function and homology information | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.912 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.912 Å | ||||||
 Authors Authors | Irimia, A. / Wilson, I.A. | ||||||
 Citation Citation | Journal: Nat.Struct.Mol.Biol. / Year: 2014 Title: Stapled HIV-1 peptides recapitulate antigenic structures and engage broadly neutralizing antibodies. Authors: Bird, G.H. / Irimia, A. / Ofek, G. / Kwong, P.D. / Wilson, I.A. / Walensky, L.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4nhc.cif.gz | 192.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4nhc.ent.gz | 153.8 KB | Display | PDB format |
| PDBx/mmJSON format | 4nhc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nh/4nhcftp://data.pdbj.org/pub/pdb/validation_reports/nh/4nhc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4nghC  2fx7S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein/peptide , 1 types, 1 molecules P
| #3: Protein/peptide |   Type: Peptide-like / Class: Unknown / Mass: 2089.566 Da / Num. of mol.: 1 / Source method: obtained synthetically Type: Peptide-like / Class: Unknown / Mass: 2089.566 Da / Num. of mol.: 1 / Source method: obtained syntheticallyDetails: SYNTHETIC; THIS SEQUENCE INCLUDES A MODIFIED FRAGMENT OF THE HIV ENVELOPE PROTEIN GP41 References: Stapled peptide (ACE)NWFNITN(DIV)LW(MK8)IKKKK |
|---|
-Antibody , 2 types, 2 molecules LH
| #1: Antibody | Mass: 23395.850 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:   Cricetulus griseus (Chinese hamster) / Strain (production host): CHO cells Cricetulus griseus (Chinese hamster) / Strain (production host): CHO cells |
|---|---|
| #2: Antibody | Mass: 23990.027 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host: Cricetulus griseus (Chinese hamster) / Strain (production host): CHO cells |
-Non-polymers , 3 types, 32 molecules 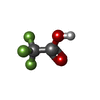


| #4: Chemical |  Mass: 114.023 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2HF3O2 Mass: 114.023 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2HF3O2#5: Chemical | ChemComp-PO4 / |  Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Compound details | THE STAPLED PEPTIDES IN THE STRUCTURE WAS OBTAINED BY INCORPORATION OF (R)-2-(((9H-FLUOREN-9-YL) ...THE STAPLED PEPTIDES IN THE STRUCTURE WAS OBTAINED BY INCORPORAT |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.12 Å3/Da / Density % sol: 60.56 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 6.2 Details: Reservoir solution: 0.2 M NaCl, 0.1 M Na/K Phosphate pH 6.2, 50% PEG 200, VAPOR DIFFUSION, SITTING DROP, temperature 22K, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 23-ID-D / Wavelength: 1 Å / Beamline: 23-ID-D / Wavelength: 1 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Dec 4, 2010 Details: K-B pair of biomorph mirrors for vertical and horizontal focusing |
| Radiation | Monochromator: double crystal monochromator, Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.91→48.95 Å / Num. all: 14400 / Num. obs: 13767 / % possible obs: 95.6 % / Observed criterion σ(I): -3 / Redundancy: 21.9 % / Biso Wilson estimate: 63.8 Å2 / Rmerge(I) obs: 0.182 / Net I/σ(I): 16.4 |
| Reflection shell | Resolution: 2.91→2.96 Å / Redundancy: 7.8 % / Rmerge(I) obs: 0.666 / Mean I/σ(I) obs: 3.1 / Num. unique all: 501 / % possible all: 70.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2FX7 Resolution: 2.912→48.95 Å / SU ML: 0.41 / σ(F): 1.34 / Phase error: 28.55 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.83 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 48.064 Å2 / ksol: 0.337 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 69 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.912→48.95 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
