 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4mwd: Trypanosoma brucei methionyl-tRNA synthetase in complex with inhi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4mwd | ||||||
|---|---|---|---|---|---|---|---|
| Title | Trypanosoma brucei methionyl-tRNA synthetase in complex with inhibitor 1-{3-[(3,5-dichlorobenzyl)amino]propyl}-3-thiophen-3-ylurea (Chem 1433) and ATP analog AMPPCP | ||||||
 Components Components | Methionyl-tRNA synthetase | ||||||
 Keywords Keywords | LIGASE/LIGASE INHIBITOR / aminoacyl-tRNA synthetase / aaRS / MetRS / parasite / protein-inhibitor complex / Rossmann fold / translation / nucleotide binding / LIGASE-LIGASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationmethionine-tRNA ligase / methionine-tRNA ligase activity / methionyl-tRNA aminoacylation / ciliary plasm / mitochondrion / ATP binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.253 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.253 Å | ||||||
 Authors Authors | Koh, C.Y. / Kim, J.E. / Wetzel, A.B. / de van der Schueren, W.J. / Shibata, S. / Liu, J. / Zhang, Z. / Fan, E. / Verlinde, C.L.M.J. / Hol, W.G.J. | ||||||
 Citation Citation | Journal: Plos Negl Trop Dis / Year: 2014 Title: Structures of Trypanosoma brucei Methionyl-tRNA Synthetase with Urea-Based Inhibitors Provide Guidance for Drug Design against Sleeping Sickness. Authors: Koh, C.Y. / Kim, J.E. / Wetzel, A.B. / de van der Schueren, W.J. / Shibata, S. / Ranade, R.M. / Liu, J. / Zhang, Z. / Gillespie, J.R. / Buckner, F.S. / Verlinde, C.L. / Fan, E. / Hol, W.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4mwd.cif.gz | 447.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4mwd.ent.gz | 362.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4mwd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mw/4mwdftp://data.pdbj.org/pub/pdb/validation_reports/mw/4mwd | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4mvwC  4mvxC  4mvyC  4mw0C  4mw1C  4mw2C  4mw4C  4mw5C  4mw6C  4mw7C  4mw9C  4mwbC  4mwcC  4mweC  4eg8S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 61538.684 Da / Num. of mol.: 2 / Fragment: UNP residues 237-773 / Mutation: K456A, K457R, E458A Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|
-Non-polymers , 7 types, 534 molecules 


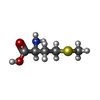

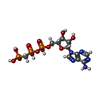

| #2: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical |  Mass: 78.133 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#4: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#5: Chemical | ChemComp-MET / |  Type: L-peptide linking / Mass: 149.211 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H11NO2S Type: L-peptide linking / Mass: 149.211 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H11NO2S#6: Chemical | ChemComp-43E / |  Mass: 358.286 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H17Cl2N3OS Mass: 358.286 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H17Cl2N3OS#7: Chemical | ChemComp-ACP / |  Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 517 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.92 Å3/Da / Density % sol: 68.65 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop Details: 2.0-2.3 M ammonium sulfate, 0.2 M sodium chloride, 0.1 M sodium cacodylate, pH 6.0-6.8, VAPOR DIFFUSION, SITTING DROP, temperature 298K PH range: 6.0-6.8 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL12-2 / Wavelength: 0.98 Å / Beamline: BL12-2 / Wavelength: 0.98 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Dec 12, 2011 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Liquid nitrogen-cooled double crystal Si(111) Protocol: SINGLE WAVELENGTH / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.253→39.967 Å / Num. all: 91992 / Num. obs: 91992 / % possible obs: 99.8 % / Redundancy: 7.8 % / Rsym value: 0.177 / Net I/σ(I): 11.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Rmerge(I) obs: 0.012 / Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4EG8 Resolution: 2.253→30 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.944 / WRfactor Rfree: 0.211 / WRfactor Rwork: 0.182 / Occupancy max: 1 / Occupancy min: 0.5 / SU B: 9.356 / SU ML: 0.118 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.177 / ESU R Free: 0.157 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 118.27 Å2 / Biso mean: 38.6552 Å2 / Biso min: 17.81 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.253→30 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
