 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4lip: PSEUDOMONAS LIPASE COMPLEXED WITH RC-(RP, SP)-DIBUTYLCARBAMOYLGLY... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4lip | ||||||
|---|---|---|---|---|---|---|---|
| Title | PSEUDOMONAS LIPASE COMPLEXED WITH RC-(RP, SP)-DIBUTYLCARBAMOYLGLYCERO-3-O-BUTYLPHOSPHONATE | ||||||
 Components Components | TRIACYL-GLYCEROL-HYDROLASE | ||||||
 Keywords Keywords | LIPASE / HYDROLASE / PSEUDOMONADACEAE / COVALENT INTERMEDIATE / TRIGLYCERIDE ANALOGUE / ENANTIOSELECTIVITY | ||||||
| Function / homology |  Function and homology information Function and homology informationtriacylglycerol lipase / triacylglycerol lipase activity / lipid catabolic process / extracellular region / metal ion binding Similarity search - Function | ||||||
| Biological species |  Burkholderia cepacia (bacteria) Burkholderia cepacia (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å | ||||||
 Authors Authors | Lang, D.A. / Dijkstra, B.W. | ||||||
 Citation Citation | Journal: Eur.J.Biochem. / Year: 1998 Title: Structural basis of the chiral selectivity of Pseudomonas cepacia lipase Authors: Lang, D.A. / Mannesse, M.L.M. / De Haas, G. / Verheij, H.M. / Dijkstra, B.W. #1: Journal: Structure / Year: 1997Title: The Open Conformation of a Pseudomonas Lipase Authors: Schrag, J.D. / Li, Y. / Cygler, M. / Lang, D. / Burgdorf, T. / Hecht, H.J. / Schmid, R. / Schomburg, D. / Rydel, T.J. / Oliver, J.D. / Strickland, L.C. / Dunaway, C.M. / Larson, S.B. / Day, J. / McPherson, A. #2: Journal: J.Bacteriol. / Year: 1991Title: Extracellular Lipase of Pseudomonas Sp. Strain Atcc 21808: Purification, Characterization, Crystallization, and Preliminary X-Ray Diffraction Data Authors: Kordel, M. / Hofmann, B. / Schomburg, D. / Schmid, R.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4lip.cif.gz | 141.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4lip.ent.gz | 107.8 KB | Display | PDB format |
| PDBx/mmJSON format | 4lip.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/li/4lipftp://data.pdbj.org/pub/pdb/validation_reports/li/4lip | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5lipC  3lipS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.9998, 0.0208, 0.001), Vector: |
-Components
| #1: Protein | Mass: 33150.766 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Burkholderia cepacia (bacteria) / Cellular location: EXTRACELLULAR / Plasmid: PHES12 / Production host: Pseudomonas sp. (bacteria) / Strain (production host): 21808 / References: UniProt: P22088, triacylglycerol lipase#2: Chemical |   Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#3: Chemical | 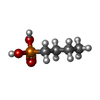  Mass: 138.102 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H11O3P Mass: 138.102 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H11O3P#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 563 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 563 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Nonpolymer details | THE STARTING MATERIAL FOR THE INHIBITOR WAS RC-(RP,SP)-1,2-DIBUTYLCARBAMOYL-GLYCERO-3-O-P- ...THE STARTING MATERIAL FOR THE INHIBITOR WAS RC-(RP,SP)-1,2-DIBUTYLCAR | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 45 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8.5 / Details: 20 % MPD, 100 MM CACL2, 0.1 M TRIS/HCL, PH 8.5 | ||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 12 ℃ / pH: 9 / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 90 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MPG/DESY, HAMBURG  / Beamline: BW6 / Wavelength: 1 / Beamline: BW6 / Wavelength: 1 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jul 1, 1996 / Details: MIRRORS |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→30 Å / Num. obs: 56937 / % possible obs: 95.3 % / Observed criterion σ(I): 0 / Redundancy: 2.46 % / Rsym value: 0.031 / Net I/σ(I): 45.8 |
| Reflection shell | Resolution: 1.75→1.83 Å / Mean I/σ(I) obs: 26.2 / Rsym value: 0.059 / % possible all: 93.9 |
| Reflection | *PLUS Num. measured all: 140279 / Rmerge(I) obs: 0.031 |
| Reflection shell | *PLUS Rmerge(I) obs: 0.059 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3LIP Resolution: 1.75→20 Å / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / σ(F): 0 Details: THE STRUCTURE WAS INITIALLY REFINED WITH STRICT NON-CRYSTALLOGRAPHIC SYMMETRY, AND THEN WITH TIGHTLY RESTRAINED NON-CRYSTALLOGRAPHIC SYMMETRY FOR ALL PARTS OF THE MOLECULE EXCEPT RESIDUES 16 ...Details: THE STRUCTURE WAS INITIALLY REFINED WITH STRICT NON-CRYSTALLOGRAPHIC SYMMETRY, AND THEN WITH TIGHTLY RESTRAINED NON-CRYSTALLOGRAPHIC SYMMETRY FOR ALL PARTS OF THE MOLECULE EXCEPT RESIDUES 16 - 28, 127 - 159, AND 218 - 224. THE CHAINS ARE NAMED D AND E, WITH MOLECULE D AS THE REFERENCE FOR SECONDARY STRUCTURE DEFINITION. DEVIATIONS FROM NON-CRYSTALLOGRAPHIC SYMMETRY CORRESPOND TO THOSE PARTS OF THE PROTEIN EXCLUDED FROM THE NCS-RESTRAINT.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 8.35 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.2 Å / Luzzati d res low obs: 8 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: PARTIAL RESTRAINT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.75→1.83 Å / Total num. of bins used: 8 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.843 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
