 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4iya: Structure of the Y250A mutant of the PANTON-VALENTINE LEUCOCIDIN ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4iya | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the Y250A mutant of the PANTON-VALENTINE LEUCOCIDIN S component from STAPHYLOCOCCUS AUREUS | ||||||
 Components Components | LukS-PV | ||||||
 Keywords Keywords | TOXIN / Staphylococcus aureus / S component leucocidin / bi-component leucotoxin / beta-barrel pore forming toxin | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Staphylococcus phage PVL (virus) Staphylococcus phage PVL (virus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å | ||||||
 Authors Authors | Maveyraud, L. / Guerin, F. / Laventie, B.J. / Prevost, G. / Mourey, L. | ||||||
 Citation Citation | Journal: Plos One / Year: 2014 Title: Residues essential for panton-valentine leukocidin s component binding to its cell receptor suggest both plasticity and adaptability in its interaction surface Authors: Laventie, B.J. / Guerin, F. / Mourey, L. / Tawk, M.Y. / Jover, E. / Maveyraud, L. / Prevost, G. #1: Journal: J.Biol.Chem. / Year: 2004Title: Crystal structure of leucotoxin S component. New insight into the staphylococcal beta-barrel pore-forming toxins Authors: Guillet, V. / Roblin, P. / Werner, S. / Coraiola, M. / Menestrina, G. / Monteil, H. / Prevost, G. / Mourey, L. #2: Journal: Acta Crystallogr.,Sect.D / Year: 2004Title: Crystallization and preliminary crystallographic data of leucotoxin S component from Staphylococcus aureus Authors: Guillet, V. / Keller, D. / Prevost, G. / Mourey, L. #3: Journal: Structure / Year: 1999Title: The structure of a Staphylococcus aureus leucocidin component (LukF-PV) reveals the fold of the water-soluble species of a family of transmembrane pore-forming toxins Authors: Pedelacq, J.D. / Maveyraud, L. / Prevost, G. / Baba-moussa, L. / Gonzalez, A. / Courcelle, M. / Shepard, W. / Monteil, H. / Samama, J.P. / Mourey, L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4iya.cif.gz | 133.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4iya.ent.gz | 103.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4iya.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/iy/4iyaftp://data.pdbj.org/pub/pdb/validation_reports/iy/4iya | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4iycC  4iytC  4izlC  4j0oC  1t5rS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 33048.375 Da / Num. of mol.: 1 / Fragment: UNP residues 29-312 / Mutation: Y250A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus phage PVL (virus) / Plasmid: pGEX-6P-1 / Production host:  | ||||
|---|---|---|---|---|---|
| #2: Chemical | 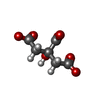  Mass: 189.100 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H5O7#3: Chemical | ChemComp-EDO / |   Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 297 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 297 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44.01 % |
|---|---|
| Crystal grow | Temperature: 285 K / Method: vapor diffusion, sitting drop / pH: 4 Details: 5% PEG 6000, 0.1M NaCitrate , pH 4.00, VAPOR DIFFUSION, SITTING DROP, temperature 285K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.948 Å / Beamline: ID14-4 / Wavelength: 0.948 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Nov 10, 2008 |
| Radiation | Monochromator: channel cut ESRF monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.948 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→30.84 Å / Num. all: 43049 / Num. obs: 43049 / % possible obs: 99.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 6.9 % / Biso Wilson estimate: 18.72 Å2 / Rmerge(I) obs: 0.067 / Rsym value: 0.067 / Net I/σ(I): 18.8 |
| Reflection shell | Resolution: 1.5→1.6 Å / Redundancy: 5.7 % / Rmerge(I) obs: 0.753 / Mean I/σ(I) obs: 3.2 / Num. unique all: 3855 / Rsym value: 0.753 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1T5R Resolution: 1.55→22.69 Å / Cor.coef. Fo:Fc: 0.9598 / Cor.coef. Fo:Fc free: 0.9537 / SU R Cruickshank DPI: 0.073 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: BUSTER TNT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.36 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.16 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.55→22.69 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.55→1.59 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 51.6669 Å / Origin y: 29.373 Å / Origin z: 21.0996 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: { A|* } |
