 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
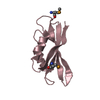
Yorodumi- PDB-4hwn: Crystal structure of the second Ig-C2 domain of human Fc-receptor... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hwn | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the second Ig-C2 domain of human Fc-receptor like A (FCRLA), Isoform 9 [NYSGRC-005836] | ||||||
 Components Components | Fc receptor-like A | ||||||
 Keywords Keywords | IMMUNE SYSTEM / FCRLA / FCRL / Ig-C2 domain / Ig superfamily / Structural genomics / PSI-biology / New York Structural Genomics Research Consortium / NYSGRC / Atoms-to-Animals: The Immune Function Network / IFN | ||||||
| Function / homology |  Function and homology information Function and homology informationplasma membrane => GO:0005886 / transmembrane signaling receptor activity / cell surface receptor signaling pathway / cell differentiation / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.006 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.006 Å | ||||||
 Authors Authors | Kumar, P.R. / Ahmed, M. / Bhosle, R. / Calarese, D. / Celikigil, A. / Chan, M.K. / Fiser, A. / Garforth, S. / Glenn, A.S. / Hillerich, B. ...Kumar, P.R. / Ahmed, M. / Bhosle, R. / Calarese, D. / Celikigil, A. / Chan, M.K. / Fiser, A. / Garforth, S. / Glenn, A.S. / Hillerich, B. / Khafizov, K. / Love, J. / Patel, H. / Rubinstein, R. / Seidel, R. / Stead, M. / Toro, R. / Nathenson, S.G. / Almo, S.C. / New York Structural Genomics Research Consortium (NYSGRC) / Atoms-to-Animals: The Immune Function Network (IFN) | ||||||
 Citation Citation | Journal: to be published Title: Crystal structure of the second Ig-C2 domain of the human Fc-receptor like A, Isoform 9 [NYSGRC-005836] Authors: Kumar, P.R. / Nathenson, S.G. / Almo, S.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hwn.cif.gz | 32.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hwn.ent.gz | 19.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4hwn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4hwn_validation.pdf.gz | 415.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4hwn_full_validation.pdf.gz | 415.4 KB | Display | |
| Data in XML | 4hwn_validation.xml.gz | 6 KB | Display | |
| Data in CIF | 4hwn_validation.cif.gz | 7.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hw/4hwnftp://data.pdbj.org/pub/pdb/validation_reports/hw/4hwn | HTTPS FTP |
-Related structure data
| Related structure data |  3rjdS S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |







-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a monomer |
-Components
| #1: Protein | Mass: 11949.231 Da / Num. of mol.: 1 / Fragment: UNP residues 169-264 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: FCRL, FCRL1, FCRLA, FCRLM1, FCRX, FREB, UNQ291/PRO329 / Plasmid: pIEX / Production host:  Trichoplusia ni (cabbage looper) / Strain (production host): Hi5 / References: UniProt: Q7L513 Trichoplusia ni (cabbage looper) / Strain (production host): Hi5 / References: UniProt: Q7L513 |
|---|---|
| #2: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 58 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 58 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.71 Å3/Da / Density % sol: 28.19 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 7 Details: Protein (20 mM Hepes, pH 7.5, 150 mM NaCl, 10% glycerol; Reservoir (0.1M Sodium Cacodylate pH 6.5, 0.2M Sodium Chloride, 2M Ammonium Sulfate); Cryoprotection (Reservoir + 2M Lithium Sulfate) ...Details: Protein (20 mM Hepes, pH 7.5, 150 mM NaCl, 10% glycerol; Reservoir (0.1M Sodium Cacodylate pH 6.5, 0.2M Sodium Chloride, 2M Ammonium Sulfate); Cryoprotection (Reservoir + 2M Lithium Sulfate), Sitting Drop Vapor Diffusion, temperature 298K, VAPOR DIFFUSION, SITTING DROP |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X29A / Wavelength: 1.075 Å / Beamline: X29A / Wavelength: 1.075 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Oct 18, 2012 / Details: MIRRORS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.075 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2→50 Å / Num. obs: 5840 / % possible obs: 99 % / Redundancy: 10.6 % / Rmerge(I) obs: 0.07 / Χ2: 0.958 / Net I/σ(I): 31.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 3RJD Resolution: 2.006→36.941 Å / Occupancy max: 1 / Occupancy min: 0.39 / SU ML: 0.23 / σ(F): 1.37 / Phase error: 24.94 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 85.55 Å2 / Biso mean: 20.0865 Å2 / Biso min: 5.06 Å2 | ||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.006→36.941 Å
| ||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 2
|
