 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2v33: High resolution crystal structure of domain III of E1 fusion glyc... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2v33 | ||||||
|---|---|---|---|---|---|---|---|
| Title | High resolution crystal structure of domain III of E1 fusion glycoprotein of Semliki Forest Virus | ||||||
 Components Components | E1 ENVELOPE GLYCOPROTEIN | ||||||
 Keywords Keywords | VIRAL PROTEIN / GLYCOPROTEIN / TRANSMEMBRANE | ||||||
| Function / homology |  Function and homology information Function and homology informationtogavirin / T=4 icosahedral viral capsid / virion assembly / small molecule binding / host cell endoplasmic reticulum / channel activity / host cell endosome / monoatomic ion transmembrane transport / clathrin-dependent endocytosis of virus by host cell / symbiont-mediated suppression of host toll-like receptor signaling pathway ...togavirin / T=4 icosahedral viral capsid / virion assembly / small molecule binding / host cell endoplasmic reticulum / channel activity / host cell endosome / monoatomic ion transmembrane transport / clathrin-dependent endocytosis of virus by host cell / symbiont-mediated suppression of host toll-like receptor signaling pathway / host cell Golgi apparatus / entry receptor-mediated virion attachment to host cell / serine-type endopeptidase activity / viral translational frameshifting / fusion of virus membrane with host endosome membrane / viral envelope / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / RNA binding Similarity search - Function | ||||||
| Biological species |   SEMLIKI FOREST VIRUS SEMLIKI FOREST VIRUS | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å | ||||||
 Authors Authors | Vaney, M.C. / Rey, F.A. | ||||||
 Citation Citation | Journal: To be Published Title: High Resolution Structure of Domain III from Class II Fusion Protein of Semliki Forest Virus Authors: Vaney, M.C. / Vigouroux, A. / Rey, F.A. #1: Journal: Structure / Year: 2006Title: Structure and Interactions at the Viral Surface of the Envelope Protein E1 of Semliki Forest Virus. Authors: Roussel, A. / Lescar, J. / Vaney, M.C. / Wengler, G. / Rey, F.A. #2: Journal: Nature / Year: 2004Title: Conformational Change and Protein-Protein Interactions of the Fusion Protein of Semliki Forest Virus. Authors: Gibbons, D.L. / Vaney, M.C. / Roussel, A. / Vigouroux, A. / Reilly, B. / Lepault, J. / Kielian, M. / Rey, F.A. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: AUTHOR PROVIDED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2v33.cif.gz | 86.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2v33.ent.gz | 67.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2v33.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v3/2v33ftp://data.pdbj.org/pub/pdb/validation_reports/v3/2v33 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1rerS S: Starting model for refinement |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 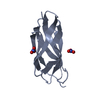
| |||||||||
| 2 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.02945, 0.9988, 0.03936), Vector: |
-Components
| #1: Protein | Mass: 9347.467 Da / Num. of mol.: 2 Fragment: DOMAIN III OF SPIKE GLYCOPROTEIN E1, RESIDUES 1107-1197 Source method: isolated from a natural source / Source: (natural) SEMLIKI FOREST VIRUS / References: UniProt: P03315#2: Chemical |   Mass: 62.005 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: NO3 Mass: 62.005 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: NO3#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 137 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 137 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 37 % / Description: NONE |
|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 6.5 Details: 25% PEG 8K, 0.2M NA ACETATE, 0.1M CACO PH 6.5, VAPOR DIFFUSION, HANGING DROP |
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.934 / Beamline: ID14-2 / Wavelength: 0.934 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Nov 4, 2005 |
| Radiation | Monochromator: DIAMOND(111), GE(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.934 Å / Relative weight: 1 |
| Reflection | Resolution: 1.55→28.57 Å / Num. obs: 31583 / % possible obs: 98.8 % / Observed criterion σ(I): 0 / Redundancy: 4.4 % / Rmerge(I) obs: 0.05 / Net I/σ(I): 23.7 |
| Reflection shell | Resolution: 1.5→1.55 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.56 / % possible all: 95.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1RER (DOMAIN III) Resolution: 1.55→28.57 Å / Cor.coef. Fo:Fc: 0.954 / Cor.coef. Fo:Fc free: 0.94 / SU B: 3.078 / SU ML: 0.051 / Cross valid method: THROUGHOUT / ESU R: 0.1 / ESU R Free: 0.083 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE FOLLOWING RESIDUES ADOPT AN ALTERNATE CONFORMATION. FOR CHAIN A, THR 300A, THR 319A, VAL 336A, SER 362A, THR 307A, THR343A, LYS 351A, ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE FOLLOWING RESIDUES ADOPT AN ALTERNATE CONFORMATION. FOR CHAIN A, THR 300A, THR 319A, VAL 336A, SER 362A, THR 307A, THR343A, LYS 351A, SER 357A. FOR CHAIN B, THR 300B, ,THR 302B,THR 305B,CYS 306B,THR 307B,LOOP PHE312B- GLY313B- -GLY314B, THR 319B,LOOP SER332B-HIS333B-SER334B-ASN 335B, ,VAL 336B,THR 338B,THR 343B,SER 357B,SER 362B,SER 371B. DISORDERED REGIONS WERE MODELED STEREOCHEMICALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.05 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.55→28.57 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|