 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4hgp: Crystal Structure of 2-keto-3-deoxyoctulosonate 8-phosphate phosp... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hgp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of 2-keto-3-deoxyoctulosonate 8-phosphate phosphohydrolase from Haemophilus influenzae in complex with transition state mimic | ||||||
 Components Components | 3-deoxy-D-manno-octulosonate 8-phosphate phosphatase KdsC | ||||||
 Keywords Keywords | HYDROLASE / Rossmann Fold / Phosphohydroylase | ||||||
| Function / homology |  Function and homology information Function and homology information3-deoxy-manno-octulosonate-8-phosphatase / 3-deoxy-manno-octulosonate-8-phosphatase activity / lipopolysaccharide biosynthetic process / metal ion binding Similarity search - Function | ||||||
| Biological species |  Haemophilus influenzae (bacteria) Haemophilus influenzae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Daughtry, K.D. / Allen, K.N. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2013 Title: Structural Basis for the Divergence of Substrate Specificity and Biological Function within HAD Phosphatases in Lipopolysaccharide and Sialic Acid Biosynthesis. Authors: Daughtry, K.D. / Huang, H. / Malashkevich, V. / Patskovsky, Y. / Liu, W. / Ramagopal, U. / Sauder, J.M. / Burley, S.K. / Almo, S.C. / Dunaway-Mariano, D. / Allen, K.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hgp.cif.gz | 53.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hgp.ent.gz | 37 KB | Display | PDB format |
| PDBx/mmJSON format | 4hgp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hg/4hgpftp://data.pdbj.org/pub/pdb/validation_reports/hg/4hgp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3mmzC  3mn1C  3n07C  4hgnC  4hgoC  4hgqC  4hgrC  1k1eS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
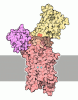
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 19455.283 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Haemophilus influenzae (bacteria) / Strain: KW20 / Gene: HI_1679 / Plasmid: PDEST17-HI1679 / Production host: References: UniProt: P45314, 3-deoxy-manno-octulosonate-8-phosphatase |
|---|---|
| #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #3: Chemical | ChemComp-VN4 / 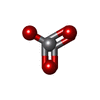  Mass: 98.940 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: VO3 Mass: 98.940 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: VO3 |
| #4: Sugar | ChemComp-KDO / 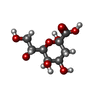  Type: D-saccharide, alpha linking / Mass: 238.192 Da / Num. of mol.: 1 Type: D-saccharide, alpha linking / Mass: 238.192 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C8H14O8 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 177 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 177 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.42 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: 100 mM Tris, 30% polyethylene glycol 3350. Crystal soaked with 20 mM NaVN4, 20 mM KDO for 3 days. Crystal dragged through Paratone prior to flash cooling, pH 8.5, VAPOR DIFFUSION, HANGING DROP, temperature 290K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR / Wavelength: 1.54 Å |
| Detector | Type: Bruker Platinum 135 / Detector: CCD / Date: Jan 14, 2010 / Details: Helios multi-layer optics |
| Radiation | Monochromator: rotating-anode / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→29.5 Å / Num. obs: 14655 / % possible obs: 95.6 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 7.9 % / Rsym value: 0.0573 / Net I/σ(I): 13.68 |
| Reflection shell | Resolution: 1.8→1.89 Å / Redundancy: 5.7 % / Mean I/σ(I) obs: 2.4 / Rsym value: 0.3813 / % possible all: 91.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1k1e Resolution: 1.8→29.463 Å / σ(F): 1.38 / Phase error: 27.98 / Stereochemistry target values: TWIN_LSQ_F
| ||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.75 Å2 | ||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→29.463 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
