 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4hfv: Crystal structure of lpg1851 protein from Legionella pneumophila ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hfv | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of lpg1851 protein from Legionella pneumophila (putative T4SS effector) | ||||||
 Components Components | Uncharacterized protein | ||||||
 Keywords Keywords | Structural Genomics / Unknown Function / PSI-Biology / Midwest Center for Structural Genomics / MCSG / effector | ||||||
| Function / homology |  Function and homology information Function and homology informationde novo design (two linked rop proteins) - #330 / Substrate of the Dot/Icm secretion system / Putative substrate of the Dot/Icm secretion system / Putative substrate of the Dot/Icm secretion system superfamily / Substrate of the Dot/Icm secretion system, putative / Methane Monooxygenase Hydroxylase; Chain G, domain 1 / de novo design (two linked rop proteins) / Helix non-globular / Special / Up-down Bundle / Mainly Alpha Similarity search - Domain/homology | ||||||
| Biological species |  Legionella pneumophila subsp. pneumophila (bacteria) Legionella pneumophila subsp. pneumophila (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.901 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.901 Å | ||||||
 Authors Authors | Michalska, K. / Xu, X. / Cui, H. / Savchenko, A. / Joachimiak, A. / Midwest Center for Structural Genomics (MCSG) | ||||||
 Citation Citation | Journal: Mol Syst Biol / Year: 2016 Title: Diverse mechanisms of metaeffector activity in an intracellular bacterial pathogen, Legionella pneumophila. Authors: Urbanus, M.L. / Quaile, A.T. / Stogios, P.J. / Morar, M. / Rao, C. / Di Leo, R. / Evdokimova, E. / Lam, M. / Oatway, C. / Cuff, M.E. / Osipiuk, J. / Michalska, K. / Nocek, B.P. / Taipale, M. ...Authors: Urbanus, M.L. / Quaile, A.T. / Stogios, P.J. / Morar, M. / Rao, C. / Di Leo, R. / Evdokimova, E. / Lam, M. / Oatway, C. / Cuff, M.E. / Osipiuk, J. / Michalska, K. / Nocek, B.P. / Taipale, M. / Savchenko, A. / Ensminger, A.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hfv.cif.gz | 98.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hfv.ent.gz | 76.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4hfv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hf/4hfvftp://data.pdbj.org/pub/pdb/validation_reports/hf/4hfv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4rxiC  4rxvC  4xa9C  5dggC C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | THE AUTHOR STATES THAT THE BIOLOGICAL UNIT OF THIS PROTEIN IS UNKNOWN. |
-Components
| #1: Protein | Mass: 22213.021 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Legionella pneumophila subsp. pneumophila (bacteria)Strain: Philadelphia 1 / Gene: lpg1851 / Plasmid: p15Tv lic / Production host: |
|---|---|
| #2: Chemical | ChemComp-CIT /   Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 |
| #3: Chemical | ChemComp-SIN / 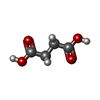  Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4 Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 197 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 197 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.51 Å3/Da / Density % sol: 50.99 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 5 Details: 0.2 M diammonium citrate, 20% PEG3350, 1/70 thermolysin, pH 5.0, VAPOR DIFFUSION, HANGING DROP, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.9794534 Å / Beamline: 19-ID / Wavelength: 0.9794534 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jul 14, 2012 / Details: mirrors |
| Radiation | Monochromator: double crystal / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9794534 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→50 Å / Num. all: 18726 / Num. obs: 18684 / % possible obs: 99.8 % / Observed criterion σ(I): -3 / Redundancy: 4.5 % / Biso Wilson estimate: 21 Å2 / Rmerge(I) obs: 0.104 / Net I/σ(I): 13.4 |
| Reflection shell | Resolution: 1.9→1.93 Å / Redundancy: 4 % / Rmerge(I) obs: 0.542 / Mean I/σ(I) obs: 2.06 / Num. unique all: 899 / % possible all: 99.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.901→27.723 Å / SU ML: 0.2 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / Phase error: 23.19 / Stereochemistry target values: ML Details: HYDROGEN ATOMS HAVE BEEN ADDED AT RIDING POSITIONS, THE PROTEIN WAS SUBJECTED TO IN SITU PROTEOLYSIS, THEREFORE THE EXACT LENGTH OF THE CRYSTALLIZED POLYPEPTIDE COULD NOT BE DETERMINED
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.901→27.723 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
