 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4h5g: Crystal structure of an amino acid ABC transporter substrate-bind... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4h5g | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of an amino acid ABC transporter substrate-binding protein from Streptococcus pneumoniae Canada MDR_19A bound to L-arginine, form 2 | ||||||
 Components Components | Amino acid ABC superfamily ATP binding cassette transporter, binding protein | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / CENTER FOR STRUCTURAL GENOMICS OF INFECTIOUS DISEASES (CSGID) / NIAID / NATIONAL INSTITUTE OF ALLERGY AND INFECTIOUS DISEASES / ALPHA AND BETA PROTEIN / PERIPLASMIC BINDING PROTEIN TYPE II FOLD / putative amino acid ABC transporter system substrate binding protein / amino acids / L-arginine / putative membrane-anchored lipoprotein | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |   Streptococcus pneumoniae (bacteria) Streptococcus pneumoniae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.78 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.78 Å | ||||||
 Authors Authors | Stogios, P.J. / Wawrzak, Z. / Kudritska, M. / Minasov, G. / Yim, V. / Savchenko, A. / Anderson, W.F. / Center for Structural Genomics of Infectious Diseases (CSGID) | ||||||
 Citation Citation | Journal: TO BE PUBLISHED Title: Crystal structure of an amino acid ABC transporter substrate-binding protein from Streptococcus pneumoniae Canada MDR_19A bound to L-arginine, form 2 Authors: Stogios, P.J. / Wawrzak, Z. / Kudritska, M. / Minasov, G. / Yim, V. / Savchenko, A. / Anderson, W.F. / Center for Structural Genomics of Infectious Diseases (CSGID) | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4h5g.cif.gz | 202.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4h5g.ent.gz | 163.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4h5g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h5/4h5gftp://data.pdbj.org/pub/pdb/validation_reports/h5/4h5g | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1wdnS S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |









-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| 2 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 26175.562 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus pneumoniae (bacteria) / Strain: Canada MDR_19A / Gene: HMPREF0837_11153, SpneCM_010100001092 / Plasmid: P15TV LIC / Production host: |
|---|
-Non-polymers , 7 types, 434 molecules 




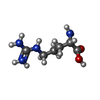

| #2: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical |  Mass: 106.120 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H10O3#4: Chemical |  Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2#5: Chemical |  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#6: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2#7: Chemical |  Type: L-peptide linking / Mass: 175.209 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15N4O2 Type: L-peptide linking / Mass: 175.209 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H15N4O2#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 419 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | THE ELECTRON DENSITY CLEARLY SUPPORTS HISTIDINE AT POSITION 136 RATHER THAN ARGININE. HOWEVER, NO ...THE ELECTRON DENSITY CLEARLY SUPPORTS HISTIDINE AT POSITION 136 RATHER THAN ARGININE. HOWEVER, NO SUITABLE SEQUENCE IN THE GENOME DATABASES WAS FOUND. HIS 136 IS ASSUMED TO BE A SEQUENCE MUTATION IN THE GENE WE WORKED WITH, RELATIVE TO THE DATABASE SEQUENCE D6ZRZ2. |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.71 Å3/Da / Density % sol: 54.67 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 0.2 M ammonium sulfate, 0.1 M sodium acetate pH 4.6, PEG 2K MME 30% (w/v), 2% tacsimate, VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-G / Wavelength: 0.97856 Å / Beamline: 21-ID-G / Wavelength: 0.97856 Å |
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Jul 1, 2012 |
| Radiation | Monochromator: C(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97856 Å / Relative weight: 1 |
| Reflection | Resolution: 1.78→91.97 Å / Num. obs: 54996 / % possible obs: 100 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -2 / Redundancy: 7.8 % / Rmerge(I) obs: 0.079 / Net I/σ(I): 17.6 |
| Reflection shell | Resolution: 1.78→1.88 Å / Redundancy: 7.8 % / Rmerge(I) obs: 0.561 / Mean I/σ(I) obs: 3.6 / Num. unique all: 61601 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1WDN Resolution: 1.78→91.97 Å / SU ML: 0.11 / Cross valid method: random / σ(F): 1.35 / Phase error: 16.85 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.78→91.97 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
