regulation of postsynaptic density protein 95 clustering / histone H3K4me1 reader activity / histone H3K14ac reader activity / positive regulation of filopodium assembly / positive regulation of dendritic spine development / modulation of excitatory postsynaptic potential / positive regulation of dendritic spine maintenance / site of DNA damage / protein localization to chromatin / negative regulation of cell migration ...regulation of postsynaptic density protein 95 clustering / histone H3K4me1 reader activity / histone H3K14ac reader activity / positive regulation of filopodium assembly / positive regulation of dendritic spine development / modulation of excitatory postsynaptic potential / positive regulation of dendritic spine maintenance / site of DNA damage / protein localization to chromatin / negative regulation of cell migration / dendritic shaft / positive regulation of transcription elongation by RNA polymerase II / double-strand break repair via homologous recombination / transcription corepressor activity / nervous system development / DNA-binding transcription factor binding / dendritic spine / protein domain specific binding / nucleolus / chromatin / negative regulation of transcription by RNA polymerase II / zinc ion binding / nucleoplasm / nucleus / cytoplasm Similarity search - Function
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.9796 Å / Relative weight: 1
Reflection
Resolution: 1.67→49.2 Å / Num. obs: 38079 / % possible obs: 99.9 % / Observed criterion σ(I): -3 / Redundancy: 6 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 12.2
Reflection shell
Resolution: 1.67→1.77 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.77 / Mean I/σ(I) obs: 2 / % possible all: 99.7
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0049
refinement
XDS
datareduction
SCALA
datascaling
SHELXD
phasing
Refinement
Method to determine structure: SAD Starting model: NONE Resolution: 1.67→49.2 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.956 / SU B: 5.168 / SU ML: 0.088 / Cross valid method: THROUGHOUT / ESU R: 0.103 / ESU R Free: 0.102 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES WITH TLS ADDED
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.21039
1900
5 %
RANDOM
Rwork
0.17589
-
-
-
obs
0.17753
36091
99.81 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj

 Assembly
Assembly


 Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn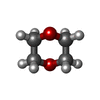
 Mass: 88.105 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H8O2
Mass: 88.105 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 18.015 Da / Num. of mol.: 333 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 333 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I04 / Wavelength: 0.9796
/ Beamline: I04 / Wavelength: 0.9796  Processing
Processing