 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4au5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the NhaA dimer, crystallised at low pH | ||||||
 Components Components | NA(+)/H(+) ANTIPORTER NHAA | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / TRANSPORTER | ||||||
| Function / homology |  Function and homology information Function and homology informationresponse to alkaline pH / sodium:proton antiporter activity / cardiolipin binding / response to salt stress / regulation of intracellular pH / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.696 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.696 Å | ||||||
 Authors Authors | Drew, D. / Lee, C. / Iwata, S. / Cameron, A.D. | ||||||
 Citation Citation | Journal: J. Gen. Physiol. / Year: 2014 Title: Crystal structure of the sodium-proton antiporter NhaA dimer and new mechanistic insights. Authors: Lee, C. / Yashiro, S. / Dotson, D.L. / Uzdavinys, P. / Iwata, S. / Sansom, M.S. / von Ballmoos, C. / Beckstein, O. / Drew, D. / Cameron, A.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4au5.cif.gz | 590.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4au5.ent.gz | 496.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4au5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/au/4au5ftp://data.pdbj.org/pub/pdb/validation_reports/au/4au5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4atvSC  1zcdS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS oper:
|
-Components
| #1: Protein | Mass: 42957.160 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Sugar | ChemComp-LMU / | 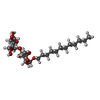  Type: D-saccharide / Mass: 510.615 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H46O11 / Comment: detergent*YM Type: D-saccharide / Mass: 510.615 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H46O11 / Comment: detergent*YM#3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.8 Å3/Da / Density % sol: 70 % / Description: NONE |
|---|---|
| Crystal grow | pH: 3.8 Details: 0.1 M SODIUM CITRATE PH 3.8, 0.1 M LITHIUM SULPHATE, 26% PEG 400 WITH 1% HEPTYL-THIOL-B-D-GLUCOSIDE AS AN ADDITIVE |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I24 / Wavelength: 0.9778 / Beamline: I24 / Wavelength: 0.9778 |
| Detector | Type: MARRESEARCH MX300 / Detector: CCD / Date: Apr 24, 2009 |
| Radiation | Monochromator: SI(III) DOUBLE CRYSTAL MONOCHOMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9778 Å / Relative weight: 1 |
| Reflection | Resolution: 3.7→30 Å / Num. obs: 34273 / % possible obs: 98.1 % / Observed criterion σ(I): -1 / Redundancy: 3.4 % / Biso Wilson estimate: 133.33 Å2 / Rmerge(I) obs: 0.1 / Net I/σ(I): 11.7 |
| Reflection shell | Resolution: 3.7→3.8 Å / Redundancy: 3 % / Rmerge(I) obs: 0.82 / Mean I/σ(I) obs: 1.2 / % possible all: 90 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1ZCD -FINAL REFINEMENT DONE AGAINST 4ATV Resolution: 3.696→29.644 Å / SU ML: 0.6 / σ(F): 1.38 / Phase error: 43.16 / Stereochemistry target values: ML Details: RESTRAINED TO 4ATV USED SECONDARY STRUCTURE AND NCS RESTRAINTS. REFINED WITH NCS RESTRAINTS AND RESTRAINED TO 4ATV. SULPHATE AND DETERGENT TENTATIVELY MODELLED IN DIMER INTERFACE AS FOR 4ATV.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.47 Å / VDW probe radii: 0.8 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 56.901 Å2 / ksol: 0.186 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 205 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.696→29.644 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
