 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4ami: Turkey beta1 adrenergic receptor with stabilising mutations and b... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ami | ||||||
|---|---|---|---|---|---|---|---|
| Title | Turkey beta1 adrenergic receptor with stabilising mutations and bound biased agonist bucindolol | ||||||
 Components Components | BETA-1 ADRENERGIC RECEPTOR | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / 7TMR BETA1-ADRENOCEPTOR / STABILISING MUTATIONS / BIASED AGONIST | ||||||
| Function / homology |  Function and homology information Function and homology informationbeta1-adrenergic receptor activity / positive regulation of heart contraction / regulation of circadian sleep/wake cycle, sleep / norepinephrine-epinephrine-mediated vasodilation involved in regulation of systemic arterial blood pressure / adenylate cyclase-activating adrenergic receptor signaling pathway / positive regulation of cardiac muscle cell apoptotic process / early endosome / positive regulation of MAPK cascade / membrane / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.2 Å | ||||||
 Authors Authors | Warne, T. / Edwards, P.C. / Leslie, A.G. / Tate, C.G. | ||||||
 Citation Citation | Journal: Structure / Year: 2012 Title: Crystal Structures of a Stabilized Beta1-Adrenoceptor Bound to the Biased Agonists Bucindolol and Carvedilol Authors: Warne, T. / Edwards, P.C. / Leslie, A.G. / Tate, C.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ami.cif.gz | 129.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ami.ent.gz | 101.1 KB | Display | PDB format |
| PDBx/mmJSON format | 4ami.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/am/4amiftp://data.pdbj.org/pub/pdb/validation_reports/am/4ami | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4amjC  2y02S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS oper:
|
-Components
| #1: Protein | Mass: 35940.777 Da / Num. of mol.: 2 / Fragment: RESIDUES 33-243,272-368 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.)  TRICHOPLUSIA NI (cabbage looper) / References: UniProt: P07700 TRICHOPLUSIA NI (cabbage looper) / References: UniProt: P07700#2: Chemical | 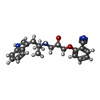  Mass: 363.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H25N3O2 Mass: 363.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H25N3O2#3: Chemical | ChemComp-2CV / 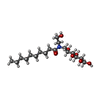  Mass: 379.489 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C18H37NO7 / Comment: detergent*YM Mass: 379.489 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C18H37NO7 / Comment: detergent*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 13 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 13 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED RESIDUE IN CHAIN A, ARG 68 TO SER ENGINEERED RESIDUE IN CHAIN A, MET 90 TO VAL ...ENGINEERED | Has protein modification | Y | Sequence details | THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE THERMOSTABILITY R68S,M90V,Y227A,A282L,F327A,F338M THE ...THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE THERMOSTAB | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.82 Å3/Da / Density % sol: 67.8 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 277 K / pH: 9 Details: 0.1 M BICINE PH 9.0, 22% PEG 600, 4 DEGREES CELSIUS. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.8726 / Beamline: ID23-2 / Wavelength: 0.8726 |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Jun 3, 2012 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8726 Å / Relative weight: 1 |
| Reflection | Resolution: 3.2→52.1 Å / Num. obs: 17466 / % possible obs: 95.9 % / Observed criterion σ(I): 2 / Redundancy: 5.3 % / Rmerge(I) obs: 0.1 / Net I/σ(I): 9.9 |
| Reflection shell | Resolution: 3.2→3.37 Å / Redundancy: 5.4 % / Rmerge(I) obs: 0.6 / Mean I/σ(I) obs: 2.5 / % possible all: 86.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2Y02 Resolution: 3.2→44.82 Å / Cor.coef. Fo:Fc: 0.91 / Cor.coef. Fo:Fc free: 0.844 / SU B: 19.89 / SU ML: 0.348 / Cross valid method: THROUGHOUT / ESU R Free: 0.49 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT. U VALUES REFINED INDIVIDUALLY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 92.661 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.2→44.82 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|