 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4ah9: Parallel screening of a low molecular weight compound library: do... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ah9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Parallel screening of a low molecular weight compound library: do differences in methodology affect hit identification | ||||||
 Components Components | INTEGRASE | ||||||
 Keywords Keywords | TRANSFERASE / FRAGMENT SCREENING | ||||||
| Function / homology |  Function and homology information Function and homology informationHIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding ...HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / viral penetration into host nucleus / host multivesicular body / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / host cell / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / symbiont-mediated suppression of host gene expression / viral translational frameshifting / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |   HUMAN IMMUNODEFICIENCY VIRUS HUMAN IMMUNODEFICIENCY VIRUS | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Wielens, J. / Heady, S.J. / Rhodes, D.I. / Mulder, R.J. / Dolezal, O. / Deadman, J.J. / Newman, J. / Chalmers, D.K. / Parker, M.W. / Peat, T.S. / Scanlon, M.J. | ||||||
 Citation Citation | Journal: J.Biomol.Screen / Year: 2013 Title: Parallel Screening of Low Molecular Weight Fragment Libraries: Do Differences in Methodology Affect Hit Identification? Authors: Wielens, J. / Headey, S.J. / Rhodes, D.I. / Mulder, R.J. / Dolezal, O. / Deadman, J.J. / Newman, J. / Chalmers, D.K. / Parker, M.W. / Peat, T.S. / Scanlon, M.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ah9.cif.gz | 87.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ah9.ent.gz | 65.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4ah9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ah/4ah9ftp://data.pdbj.org/pub/pdb/validation_reports/ah/4ah9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3vq4C  3vq5C  3vq6C  3vq7C  3vq8C  3vq9C  3vqaC  3vqbC  3vqcC  3vqdC  3vqeC  3vqpC  3vqqC  4ahrC  4ahsC  4ahtC  4ahuC  4ahvC  1bi4S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 20044.672 Da / Num. of mol.: 2 / Fragment: CATALYTIC CORE DOMAIN, RESIDUES 1197-1359 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HUMAN IMMUNODEFICIENCY VIRUS / Strain: TYPE 1 / Production host:  References: UniProt: P12497, nicotinamide-nucleotide adenylyltransferase |
|---|
-Non-polymers , 7 types, 216 molecules 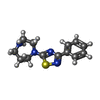




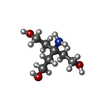

| #2: Chemical |  Mass: 260.358 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C13H16N4S Mass: 260.358 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C13H16N4S#3: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C2H6O2#5: Chemical | ChemComp-CL /  Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl#6: Chemical |  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#7: Chemical |  Mass: 163.215 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H17NO3 / Comment: pH buffer*YM Mass: 163.215 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H17NO3 / Comment: pH buffer*YM#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 187 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Compound details | ENGINEERED RESIDUE IN CHAIN A, CYS 1203 TO SER ENGINEERED RESIDUE IN CHAIN A, PHE 1286 TO ASP ...ENGINEERED |
|---|---|
| Nonpolymer details | LIGAND (0MB): FRAGMENT FROM MAYBRIDGE LIBRARY |
| Sequence details | N-TERMINAL HIS-TAG AND C56S, F139D, F185H |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 20 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.39 Å3/Da / Density % sol: 48.6 % / Description: NONE |
|---|---|
| Crystal grow | pH: 5.5 Details: 100 MM SODIUM ACETATE PH 5.0 TO 5.5, 1.2 M TO 1.5 M AMMONIUM SULFATE. THE PROTEIN WAS IN 40 MM TRIS PH 8.0, 250 MM NACL, 30 MM MGCL2, 5 MM DTT. FRAGMENTS WERE SOAKED INTO PREFORMED CRYSTALS ...Details: 100 MM SODIUM ACETATE PH 5.0 TO 5.5, 1.2 M TO 1.5 M AMMONIUM SULFATE. THE PROTEIN WAS IN 40 MM TRIS PH 8.0, 250 MM NACL, 30 MM MGCL2, 5 MM DTT. FRAGMENTS WERE SOAKED INTO PREFORMED CRYSTALS 24-48 HOURS PRIOR TO DATA COLLECTION. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Australian Synchrotron  / Beamline: MX1 / Wavelength: 0.95362 / Beamline: MX1 / Wavelength: 0.95362 |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Mar 20, 2008 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.95362 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→45 Å / Num. obs: 40414 / % possible obs: 98.8 % / Observed criterion σ(I): 1 / Redundancy: 5.3 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 14.2 |
| Reflection shell | Resolution: 1.7→1.79 Å / Redundancy: 3.6 % / Rmerge(I) obs: 0.75 / Mean I/σ(I) obs: 1.7 / % possible all: 92.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1BI4 Resolution: 1.7→61.08 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.944 / SU B: 1.861 / SU ML: 0.062 / Cross valid method: THROUGHOUT / ESU R: 0.094 / ESU R Free: 0.099 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT. U VALUES REFINED INDIVIDUALLY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.925 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→61.08 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|