 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4a5w | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of C5b6 | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | IMMUNE SYSTEM / IMMUNITY / MEMBRANE ATTACK COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationTerminal pathway of complement / membrane attack complex / positive regulation of activation of membrane attack complex / Activation of C3 and C5 / complement activation, GZMK pathway / negative regulation of macrophage chemotaxis / other organism cell membrane / complement activation, alternative pathway / complement activation / chemokine activity ...Terminal pathway of complement / membrane attack complex / positive regulation of activation of membrane attack complex / Activation of C3 and C5 / complement activation, GZMK pathway / negative regulation of macrophage chemotaxis / other organism cell membrane / complement activation, alternative pathway / complement activation / chemokine activity / endopeptidase inhibitor activity / complement activation, classical pathway / positive regulation of vascular endothelial growth factor production / positive regulation of chemokine production / Regulation of Complement cascade / Peptide ligand-binding receptors / transmembrane transport / positive regulation of immune response / chemotaxis / positive regulation of angiogenesis / killing of cells of another organism / G alpha (i) signalling events / in utero embryonic development / cell surface receptor signaling pathway / G protein-coupled receptor signaling pathway / inflammatory response / signaling receptor binding / : / extracellular exosome / extracellular region / plasma membrane Similarity search - Function | |||||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.5 Å | |||||||||
 Authors Authors | Hadders, M.A. / Bubeck, D. / Forneris, F. / Pangburn, M. / Llorca, O. / Lea, S.M. / Gros, P. | |||||||||
 Citation Citation | Journal: Cell Rep / Year: 2012 Title: Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Authors: Michael A Hadders / Doryen Bubeck / Pietro Roversi / Svetlana Hakobyan / Federico Forneris / B Paul Morgan / Michael K Pangburn / Oscar Llorca / Susan M Lea / Piet Gros /  Abstract: Activation of the complement system results in formation of membrane attack complexes (MACs), pores that disrupt lipid bilayers and lyse bacteria and other pathogens. Here, we present the crystal ...Activation of the complement system results in formation of membrane attack complexes (MACs), pores that disrupt lipid bilayers and lyse bacteria and other pathogens. Here, we present the crystal structure of the first assembly intermediate, C5b6, together with a cryo-electron microscopy reconstruction of a soluble, regulated form of the pore, sC5b9. Cleavage of C5 to C5b results in marked conformational changes, distinct from those observed in the homologous C3-to-C3b transition. C6 captures this conformation, which is preserved in the larger sC5b9 assembly. Together with antibody labeling, these structures reveal that complement components associate through sideways alignment of the central MAC-perforin (MACPF) domains, resulting in a C5b6-C7-C8β-C8α-C9 arc. Soluble regulatory proteins below the arc indicate a potential dual mechanism in protection from pore formation. These results provide a structural framework for understanding MAC pore formation and regulation, processes important for fighting infections and preventing complement-mediated tissue damage. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4a5w.cif.gz | 990.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4a5w.ent.gz | 821.1 KB | Display | PDB format |
| PDBx/mmJSON format | 4a5w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a5/4a5wftp://data.pdbj.org/pub/pdb/validation_reports/a5/4a5w | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 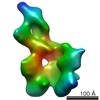 1991C  2ok5S  3cu7S  3ojyS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
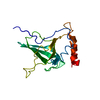
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 177707.391 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) HOMO SAPIENS (human) / Tissue: BLOOD / References: UniProt: P01031 |
|---|---|
| #2: Protein | Mass: 102541.312 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) HOMO SAPIENS (human) / Tissue: BLOOD / References: UniProt: P13671 |
-Sugars , 3 types, 8 molecules 
| #3: Sugar | ChemComp-NAG /  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 4 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #5: Sugar | ChemComp-FUC / |  Type: L-saccharide, alpha linking / Mass: 164.156 Da / Num. of mol.: 1 Type: L-saccharide, alpha linking / Mass: 164.156 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C6H12O5 #6: Sugar |  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 3 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 3Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
|---|
-Non-polymers , 1 types, 1 molecules 
| #4: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.25 Å3/Da / Density % sol: 71 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.8 / Details: 167 MM HEPES-NAOH PH7.8 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.97623 / Beamline: ID29 / Wavelength: 0.97623 |
| Detector | Type: ADSC CCD / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97623 Å / Relative weight: 1 |
| Reflection | Resolution: 3.5→50 Å / Num. obs: 59023 / % possible obs: 95.8 % / Observed criterion σ(I): 2 / Redundancy: 3.1 % / Biso Wilson estimate: 98.41 Å2 / Rmerge(I) obs: 0.12 / Net I/σ(I): 8.4 |
| Reflection shell | Resolution: 3.9→4 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.81 / Mean I/σ(I) obs: 1.7 / % possible all: 97.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRIES 3CU7, 3OJY, 2OK5 Resolution: 3.5→46.19 Å / Cor.coef. Fo:Fc: 0.8931 / Cor.coef. Fo:Fc free: 0.8864 / Cross valid method: THROUGHOUT / σ(F): 0 Details: IDEAL-DIST CONTACT TERM CONTACT SETUP. RESIDUE TYPES WITHOUT CCP4 ATOM TYPE IN LIBRARY=NAG CA FUC BMA. NUMBER OF ATOMS WITH PROPER CCP4 ATOM TYPE=18928. NUMBER WITH APPROX DEFAULT CCP4 ATOM ...Details: IDEAL-DIST CONTACT TERM CONTACT SETUP. RESIDUE TYPES WITHOUT CCP4 ATOM TYPE IN LIBRARY=NAG CA FUC BMA. NUMBER OF ATOMS WITH PROPER CCP4 ATOM TYPE=18928. NUMBER WITH APPROX DEFAULT CCP4 ATOM TYPE=99. NUMBER TREATED BY BAD NON- BONDED CONTACTS=1.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 157.24 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 1.207 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.5→46.19 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.5→3.59 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
