- PDB-3zq5: Structure of the Y263F mutant of the cyanobacterial phytochrome Cph1 -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 3zq5
Title
Structure of the Y263F mutant of the cyanobacterial phytochrome Cph1
Components
PHYTOCHROME-LIKE PROTEIN CPH1
Keywords
TRANSFERASE / ARGININE FINGER / TANDEM GAF DOMAIN / RECEPTOR / PAS DOMAIN / CHROMOPHORE / SENSORY TRANSDUCTION / PHOTORECEPTOR PROTEIN / BILIN-LIKE CHROMOPHORE / PHYTOCHROME / PHOTORECEPTOR
Function / homology
Function and homology information
red or far-red light photoreceptor activity / red, far-red light phototransduction / protein histidine kinase activity / detection of visible light / phosphorelay sensor kinase activity / histidine kinase / phosphorelay signal transduction system / regulation of DNA-templated transcription / ATP binding / identical protein binding Similarity search - Function
: / PHY domain / Phytochrome / GAF domain / Phytochrome, PHY domain superfamily / Phytochrome, central region / PAS fold-2 / PAS fold / Phytochrome region / Phytochrome chromophore attachment domain ...: / PHY domain / Phytochrome / GAF domain / Phytochrome, PHY domain superfamily / Phytochrome, central region / PAS fold-2 / PAS fold / Phytochrome region / Phytochrome chromophore attachment domain / Phytochrome chromophore attachment site domain profile. / GAF domain profile. / His Kinase A (phospho-acceptor) domain / His Kinase A (phosphoacceptor) domain / Signal transduction histidine kinase, dimerisation/phosphoacceptor domain / PAS domain / Signal transduction histidine kinase, dimerisation/phosphoacceptor domain superfamily / Histidine kinase domain / Histidine kinase domain profile. / GAF domain / Domain present in phytochromes and cGMP-specific phosphodiesterases. / GAF domain / GAF-like domain superfamily / Beta-Lactamase / PAS domain / Histidine kinase-, DNA gyrase B-, and HSP90-like ATPase / PAS domain / PAS domain superfamily / Histidine kinase-like ATPases / Histidine kinase/HSP90-like ATPase / Histidine kinase/HSP90-like ATPase superfamily / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
Resolution: 1.95→33.86 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.946 / SU B: 7.676 / SU ML: 0.096 / Cross valid method: THROUGHOUT / ESU R: 0.129 / ESU R Free: 0.129 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES, RESIDUAL ONLY
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.21765
2662
5 %
RANDOM
Rwork
0.17364
-
-
-
obs
0.17578
50593
99.32 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj


 Assembly
Assembly

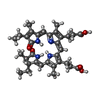





 Mass: 588.694 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H40N4O6
Mass: 588.694 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H40N4O6 Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2
Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Sample preparation
Sample preparation / Beamline: ID14-2 / Wavelength: 0.933
/ Beamline: ID14-2 / Wavelength: 0.933  Processing
Processing