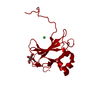
 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 3zmn | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | VP17, a capsid protein of bacteriophage P23-77 | ||||||
 要素 要素 | (VP17) x 2 | ||||||
 キーワード キーワード | VIRAL PROTEIN | ||||||
| 機能・相同性 | Jelly Rolls - #1170 / Immunoglobulin-like - #3410 / : / Large MCP VP17 central beta-barrel / Jelly Rolls / Immunoglobulin-like / Sandwich / Mainly Beta / VP17 機能・相同性情報 機能・相同性情報 | ||||||
| 生物種 |   THERMUS PHAGE P23-77 (ファージ) THERMUS PHAGE P23-77 (ファージ) | ||||||
| 手法 |  X線回折 / シンクロトロン / 単一同系置換・異常分散 / 解像度: 2.26 Å X線回折 / シンクロトロン / 単一同系置換・異常分散 / 解像度: 2.26 Å | ||||||
 データ登録者 データ登録者 | Rissanen, I. / Grimes, J.M. / Pawlowski, A. / Mantynen, S. / Harlos, K. / Bamford, J.K.H. / Stuart, D.I. | ||||||
 引用 引用 | ジャーナル: Structure / 年: 2013 タイトル: Bacteriophage P23-77 Capsid Protein Structures Reveal the Archetype of an Ancient Branch from a Major Virus Lineage. 著者: Rissanen, I. / Grimes, J.M. / Pawlowski, A. / Mantynen, S. / Harlos, K. / Bamford, J.K.H. / Stuart, D.I. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 3zmn.cif.gz | 202 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb3zmn.ent.gz | 163.8 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 3zmn.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 3zmn_validation.pdf.gz | 443 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 3zmn_full_validation.pdf.gz | 446.1 KB | 表示 | |
| XML形式データ | 3zmn_validation.xml.gz | 22.8 KB | 表示 | |
| CIF形式データ | 3zmn_validation.cif.gz | 33.8 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/zm/3zmnftp://data.pdbj.org/pub/pdb/validation_reports/zm/3zmn | HTTPS FTP |
-関連構造データ













-リンク
 PDBj
PDBj- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| 単位格子 |
| ||||||||
| Components on special symmetry positions |
|
-要素
| #1: タンパク質 | 分子量: 31875.924 Da / 分子数: 2 / 由来タイプ: 組換発現 詳細: FOR LOOPS AT 220-225 (CHAIN A) AND 221-226 (CHAIN B), POORLY ORDERED BUT CLEARLY VISIBLE POLYPEPTIDE CHAIN WAS MODELLED AS POLYALANINE. 由来: (組換発現) THERMUS PHAGE P23-77 (ファージ) / プラスミド: PIR1 (ORF17/PET22B) / 発現宿主:  #2: タンパク質・ペプチド | 分子量: 698.854 Da / 分子数: 2 / 由来タイプ: 組換発現 詳細: CHAINS C AND D ARE FRAGMENTS OF THE DISORDERED TERMINI, MODELLED AS UNK. 由来: (組換発現) THERMUS PHAGE P23-77 (ファージ) / プラスミド: PIR1 (ORF17/PET22B) / 発現宿主: #3: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 424 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 424 / 由来タイプ: 天然 / 式: H2O配列の詳細 | CERTAIN RESIDUES WERE MODELLED WITH TRUNCATED SIDECHAINS. THESE INCLUDE CHAIN A 220-225, CHAIN B ...CERTAIN RESIDUES WERE MODELLED WITH TRUNCATED SIDECHAINS | |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 3.04 Å3/Da / 溶媒含有率: 59.6 % / 解説: NONE |
|---|---|
| 結晶化 | 手法: 蒸気拡散法, シッティングドロップ法 / pH: 7 詳細: SITTING DROP VAPOUR DIFFUSION SYSTEM, MICROLITER DROPS OF PROTEIN(2-3 MG/ML IN 20 MM TRIS-BUFFER PH 7.4) MIXED 1:1 WITH SOLUTION CONSISTING OF 1.9 M SODIUM FORMATE AND 0.1 M BIS-TRIS BUFFER PH 7. |
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: Diamond  / ビームライン: I04 / 波長: 1 / ビームライン: I04 / 波長: 1 |
| 検出器 | タイプ: ADSC CCD / 検出器: CCD / 日付: 2010年6月26日 |
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1 Å / 相対比: 1 |
| 反射 | 解像度: 2.26→59.7 Å / Num. obs: 37893 / % possible obs: 100 % / 冗長度: 35.5 % / Biso Wilson estimate: 49.88 Å2 / Rmerge(I) obs: 0.08 / Net I/σ(I): 38.6 |
| 反射 シェル | 解像度: 2.26→2.32 Å / 冗長度: 36.6 % / Rmerge(I) obs: 0.92 / Mean I/σ(I) obs: 5.3 / % possible all: 100 |
- 解析
解析
| ソフトウェア |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 単一同系置換・異常分散 開始モデル: NONE 解像度: 2.26→43.94 Å / Cor.coef. Fo:Fc: 0.9393 / Cor.coef. Fo:Fc free: 0.9256 / SU R Cruickshank DPI: 0.181 / 交差検証法: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.2 / SU Rfree Blow DPI: 0.165 / SU Rfree Cruickshank DPI: 0.158 詳細: RESIDUES 1-40 AND 274-291 ARE DISORDERED IN CHAIN A. RESIDUES 1-42 AND 274-291 ARE DISORDERED IN CHAIN B. CHAINS C AND D ARE FRAGMENTS OF THE DISORDERED TERMINI AND MODELLED AS UNK. IT IS ...詳細: RESIDUES 1-40 AND 274-291 ARE DISORDERED IN CHAIN A. RESIDUES 1-42 AND 274-291 ARE DISORDERED IN CHAIN B. CHAINS C AND D ARE FRAGMENTS OF THE DISORDERED TERMINI AND MODELLED AS UNK. IT IS UNCLEAR WHICH OF CHAINS (A OR B) THESE POLYPEPTIDE FRAGMENTS BELONG TO.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 51.44 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.274 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.26→43.94 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2.26→2.32 Å / Total num. of bins used: 19
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 TLS | 手法: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 TLSグループ |
|
