 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3zmn | ||||||
|---|---|---|---|---|---|---|---|
| Title | VP17, a capsid protein of bacteriophage P23-77 | ||||||
 Components Components | (VP17) x 2 | ||||||
 Keywords Keywords | VIRAL PROTEIN | ||||||
| Function / homology | Jelly Rolls - #1170 / Immunoglobulin-like - #3410 / : / Large MCP VP17 central beta-barrel / Jelly Rolls / Immunoglobulin-like / Sandwich / Mainly Beta / VP17 Function and homology information Function and homology information | ||||||
| Biological species |   THERMUS PHAGE P23-77 (virus) THERMUS PHAGE P23-77 (virus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 2.26 Å X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 2.26 Å | ||||||
 Authors Authors | Rissanen, I. / Grimes, J.M. / Pawlowski, A. / Mantynen, S. / Harlos, K. / Bamford, J.K.H. / Stuart, D.I. | ||||||
 Citation Citation | Journal: Structure / Year: 2013 Title: Bacteriophage P23-77 Capsid Protein Structures Reveal the Archetype of an Ancient Branch from a Major Virus Lineage. Authors: Rissanen, I. / Grimes, J.M. / Pawlowski, A. / Mantynen, S. / Harlos, K. / Bamford, J.K.H. / Stuart, D.I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3zmn.cif.gz | 202 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3zmn.ent.gz | 163.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3zmn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zm/3zmnftp://data.pdbj.org/pub/pdb/validation_reports/zm/3zmn | HTTPS FTP |
|---|
-Related structure data

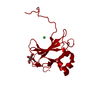












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 31875.924 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: FOR LOOPS AT 220-225 (CHAIN A) AND 221-226 (CHAIN B), POORLY ORDERED BUT CLEARLY VISIBLE POLYPEPTIDE CHAIN WAS MODELLED AS POLYALANINE. Source: (gene. exp.) THERMUS PHAGE P23-77 (virus) / Plasmid: PIR1 (ORF17/PET22B) / Production host:  #2: Protein/peptide | Mass: 698.854 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: CHAINS C AND D ARE FRAGMENTS OF THE DISORDERED TERMINI, MODELLED AS UNK. Source: (gene. exp.) THERMUS PHAGE P23-77 (virus) / Plasmid: PIR1 (ORF17/PET22B) / Production host: #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 424 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 424 / Source method: isolated from a natural source / Formula: H2OSequence details | CERTAIN RESIDUES WERE MODELLED WITH TRUNCATED SIDECHAINS. THESE INCLUDE CHAIN A 220-225, CHAIN B ...CERTAIN RESIDUES WERE MODELLED WITH TRUNCATED SIDECHAINS | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.04 Å3/Da / Density % sol: 59.6 % / Description: NONE |
|---|---|
| Crystal grow | Method: vapor diffusion, sitting drop / pH: 7 Details: SITTING DROP VAPOUR DIFFUSION SYSTEM, MICROLITER DROPS OF PROTEIN(2-3 MG/ML IN 20 MM TRIS-BUFFER PH 7.4) MIXED 1:1 WITH SOLUTION CONSISTING OF 1.9 M SODIUM FORMATE AND 0.1 M BIS-TRIS BUFFER PH 7. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 1 / Beamline: I04 / Wavelength: 1 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jun 26, 2010 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.26→59.7 Å / Num. obs: 37893 / % possible obs: 100 % / Redundancy: 35.5 % / Biso Wilson estimate: 49.88 Å2 / Rmerge(I) obs: 0.08 / Net I/σ(I): 38.6 |
| Reflection shell | Resolution: 2.26→2.32 Å / Redundancy: 36.6 % / Rmerge(I) obs: 0.92 / Mean I/σ(I) obs: 5.3 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIRAS Starting model: NONE Resolution: 2.26→43.94 Å / Cor.coef. Fo:Fc: 0.9393 / Cor.coef. Fo:Fc free: 0.9256 / SU R Cruickshank DPI: 0.181 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.2 / SU Rfree Blow DPI: 0.165 / SU Rfree Cruickshank DPI: 0.158 Details: RESIDUES 1-40 AND 274-291 ARE DISORDERED IN CHAIN A. RESIDUES 1-42 AND 274-291 ARE DISORDERED IN CHAIN B. CHAINS C AND D ARE FRAGMENTS OF THE DISORDERED TERMINI AND MODELLED AS UNK. IT IS ...Details: RESIDUES 1-40 AND 274-291 ARE DISORDERED IN CHAIN A. RESIDUES 1-42 AND 274-291 ARE DISORDERED IN CHAIN B. CHAINS C AND D ARE FRAGMENTS OF THE DISORDERED TERMINI AND MODELLED AS UNK. IT IS UNCLEAR WHICH OF CHAINS (A OR B) THESE POLYPEPTIDE FRAGMENTS BELONG TO.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 51.44 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.274 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.26→43.94 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.26→2.32 Å / Total num. of bins used: 19
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
