| Entry | Database: PDB / ID: 3wvm
|
|---|
| Title | The 0.88 angstrom X-ray structure of the human heart fatty acid-binding protein complexed with stearic acid |
|---|
 Components Components | Fatty acid-binding protein, heart |
|---|
 Keywords Keywords | LIPID BINDING PROTEIN / antiparallel beta barrel |
|---|
| Function / homology |  Function and homology information Function and homology information
icosatetraenoic acid binding / positive regulation of long-chain fatty acid import into cell / regulation of phosphatidylcholine biosynthetic process / regulation of fatty acid oxidation / oleic acid binding / positive regulation of phospholipid biosynthetic process / intracellular lipid transport / response to fatty acid / phospholipid homeostasis / long-chain fatty acid transmembrane transporter activity ...icosatetraenoic acid binding / positive regulation of long-chain fatty acid import into cell / regulation of phosphatidylcholine biosynthetic process / regulation of fatty acid oxidation / oleic acid binding / positive regulation of phospholipid biosynthetic process / intracellular lipid transport / response to fatty acid / phospholipid homeostasis / long-chain fatty acid transmembrane transporter activity / long-chain fatty acid binding / Triglyceride catabolism / sarcoplasm / long-chain fatty acid transport / cytoskeletal protein binding / brown fat cell differentiation / cholesterol homeostasis / fatty acid metabolic process / response to insulin / response to xenobiotic stimulus / negative regulation of cell population proliferation / : / extracellular exosome / nucleus / cytosolSimilarity search - Function Cytosolic fatty-acid binding proteins signature. / Intracellular lipid binding protein / Cytosolic fatty-acid binding / Lipocalin / cytosolic fatty-acid binding protein family / Lipocalin/cytosolic fatty-acid binding domain / Calycin beta-barrel core domain / Calycin / Lipocalin / Beta Barrel / Mainly BetaSimilarity search - Domain/homology |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 0.88 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 0.88 Å |
|---|
 Authors Authors | Sugiyama, S. / Matsuoka, S. / Mizohata, E. / Matsuoka, D. / Ishida, H. / Hirose, M. / Kakinouchi, K. / Hara, T. / Matsumura, H. / Murakami, S. ...Sugiyama, S. / Matsuoka, S. / Mizohata, E. / Matsuoka, D. / Ishida, H. / Hirose, M. / Kakinouchi, K. / Hara, T. / Matsumura, H. / Murakami, S. / Inoue, T. / Murata, M. |
|---|
 Citation Citation | Journal: Angew.Chem.Int.Ed.Engl. / Year: 2015
Title: Water-mediated recognition of simple alkyl chains by heart-type fatty-acid-binding protein.
Authors: Matsuoka, S. / Sugiyama, S. / Matsuoka, D. / Hirose, M. / Lethu, S. / Ano, H. / Hara, T. / Ichihara, O. / Kimura, S.R. / Murakami, S. / Ishida, H. / Mizohata, E. / Inoue, T. / Murata, M. |
|---|
| History | | Deposition | May 25, 2014 | Deposition site: PDBJ / Processing site: PDBJ |
|---|
| Revision 1.0 | Jan 28, 2015 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Aug 24, 2022 | Group: Database references / Derived calculations / Category: citation / database_2 / struct_site
Item: _citation.journal_volume / _citation.page_first ..._citation.journal_volume / _citation.page_first / _citation.page_last / _citation.title / _citation.year / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
| Revision 1.2 | May 29, 2024 | Group: Data collection / Category: chem_comp_atom / chem_comp_bond |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














 PDBj
PDBj


 Assembly
Assembly


 Mass: 284.477 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H36O2
Mass: 284.477 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H36O2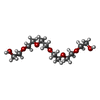
 Mass: 282.331 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM
Mass: 282.331 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM Mass: 18.015 Da / Num. of mol.: 174 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 174 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL44XU / Wavelength: 0.8 Å
/ Beamline: BL44XU / Wavelength: 0.8 Å Processing
Processing