 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
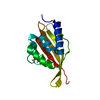
Yorodumi- PDB-3wsr: Crystal structure of CLEC-2 in complex with O-glycosylated podoplanin -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3wsr | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of CLEC-2 in complex with O-glycosylated podoplanin | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / C-type lectin fold / Cell surface receptor / Podoplanin / O-glycosylated / Extracellular region | |||||||||
| Function / homology |  Function and homology information Function and homology informationregulation of myofibroblast contraction / lymphatic endothelial cell fate commitment / actin-mediated cell contraction / leading edge of lamellipodium / regulation of substrate adhesion-dependent cell spreading / chemokine binding / positive regulation of extracellular matrix disassembly / Specification of primordial germ cells / lymphangiogenesis / regulation of lamellipodium morphogenesis ...regulation of myofibroblast contraction / lymphatic endothelial cell fate commitment / actin-mediated cell contraction / leading edge of lamellipodium / regulation of substrate adhesion-dependent cell spreading / chemokine binding / positive regulation of extracellular matrix disassembly / Specification of primordial germ cells / lymphangiogenesis / regulation of lamellipodium morphogenesis / tetraspanin-enriched microdomain / platelet formation / positive regulation of platelet aggregation / filopodium membrane / wound healing, spreading of cells / microvillus membrane / lamellipodium membrane / lymph node development / positive regulation of epithelial to mesenchymal transition / Rho protein signal transduction / GPVI-mediated activation cascade / ruffle / cell projection / lung development / filopodium / Heme signaling / defense response / platelet activation / ruffle membrane / cell junction / transmembrane signaling receptor activity / regulation of cell shape / cell migration / lamellipodium / carbohydrate binding / protein-folding chaperone binding / cytoplasmic vesicle / basolateral plasma membrane / cell surface receptor signaling pathway / cell adhesion / apical plasma membrane / positive regulation of cell migration / membrane raft / negative regulation of cell population proliferation / signaling receptor binding / negative regulation of apoptotic process / cell surface / signal transduction / mitochondrion / membrane / plasma membrane / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.91 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.91 Å | |||||||||
 Authors Authors | Nagae, M. / Morita-Matsumoto, K. / Kato, M. / Kato-Kaneko, M. / Kato, Y. / Yamaguchi, Y. | |||||||||
 Citation Citation | Journal: Structure / Year: 2014 Title: A Platform of C-type Lectin-like Receptor CLEC-2 for Binding O-Glycosylated Podoplanin and Nonglycosylated Rhodocytin Authors: Nagae, M. / Morita-Matsumoto, K. / Kato, M. / Kato-Kaneko, M. / Kato, Y. / Yamaguchi, Y. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3wsr.cif.gz | 71.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3wsr.ent.gz | 52.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3wsr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ws/3wsrftp://data.pdbj.org/pub/pdb/validation_reports/ws/3wsr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3wwkC  2c6uS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 15157.050 Da / Num. of mol.: 2 / Fragment: CLEC-2, UNP residues 96-221 / Mutation: C99S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CLEC1B, CLEC2, UNQ721/PRO1384 / Plasmid: pCold / Production host:  #2: Protein/peptide | Mass: 1703.846 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) Homo sapiens (human) / References: UniProt: Q86YL7#3: Polysaccharide | Source method: isolated from a genetically manipulated source #4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 36 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 36 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.06 Å3/Da / Density % sol: 40.25 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 0.1 M Tris-HCl (pH 8.5), 25% (w/v) polyethylene glycol (PEG) 3350, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 95 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory  / Beamline: AR-NW12A / Wavelength: 1 Å / Beamline: AR-NW12A / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: Feb 4, 2014 / Details: Monochrometer |
| Radiation | Monochromator: Si(111) double crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→100 Å / Num. obs: 21375 / % possible obs: 100 % / Observed criterion σ(I): -3 / Redundancy: 4.1 % / Rsym value: 0.072 / Net I/σ(I): 24.3 |
| Reflection shell | Resolution: 1.9→1.93 Å / Redundancy: 4.1 % / Mean I/σ(I) obs: 3.3 / Num. unique all: 1048 / Rsym value: 0.495 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB code 2C6U Resolution: 1.91→50.13 Å / Cor.coef. Fo:Fc: 0.937 / Cor.coef. Fo:Fc free: 0.908 / SU B: 4.343 / SU ML: 0.127 / Cross valid method: THROUGHOUT / ESU R: 0.191 / ESU R Free: 0.169 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.01 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.91→50.13 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|