 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3ro7: Crystal Structure of Mouse Apolipoprotein A-I Binding Protein in ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3ro7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Mouse Apolipoprotein A-I Binding Protein in Complex with Thymine. | ||||||
 Components Components | Apolipoprotein A-I-binding protein | ||||||
 Keywords Keywords | PROTEIN BINDING / ROSSMANN FOLD | ||||||
| Function / homology |  Function and homology information Function and homology informationmembrane raft distribution / Nicotinate metabolism / NAD(P)H-hydrate epimerase / NAD(P)HX epimerase activity / metabolite repair / nicotinamide nucleotide metabolic process / regulation of cholesterol efflux / sprouting angiogenesis / lipid transport / negative regulation of angiogenesis ...membrane raft distribution / Nicotinate metabolism / NAD(P)H-hydrate epimerase / NAD(P)HX epimerase activity / metabolite repair / nicotinamide nucleotide metabolic process / regulation of cholesterol efflux / sprouting angiogenesis / lipid transport / negative regulation of angiogenesis / cell body / cilium / nucleotide binding / mitochondrion / : / extracellular region / metal ion binding / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Shumilin, I.A. / Jha, K.N. / Cymborowski, M. / Herr, J.C. / Minor, W. | ||||||
 Citation Citation | Journal: Structure / Year: 2012 Title: Identification of unknown protein function using metabolite cocktail screening. Authors: Shumilin, I.A. / Cymborowski, M. / Chertihin, O. / Jha, K.N. / Herr, J.C. / Lesley, S.A. / Joachimiak, A. / Minor, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3ro7.cif.gz | 105.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3ro7.ent.gz | 82 KB | Display | PDB format |
| PDBx/mmJSON format | 3ro7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ro/3ro7ftp://data.pdbj.org/pub/pdb/validation_reports/ro/3ro7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3rnoC  3roeC  3rogC  3roxC  3rozC  3rphC  3rpzC  3rq2C  3rq5C  3rq6C  3rq8C  3rqhC  3rqqC  3rqxC  3rrbC  3rreC  3rrfC  3rrjC  3rs8C  3rs9C  3rsfC  3rsgC  3rsqC  3rssC  3rt7C  3rt9C  3rtaC  3rtbC  3rtcC  3rtdC  3rteC  3rtgC  3ru2C  3ru3C  2o8nS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | x 6
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 29872.963 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Chemical | ChemComp-TDR / 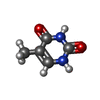  Mass: 126.113 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H6N2O2 Mass: 126.113 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H6N2O2 |
| #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 27 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 27 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.81 Å3/Da / Density % sol: 56.26 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 4.6 Details: 0.1 M SODIUM ACETATE, 1.5 M AMMONIUM SULFATE, PH 4.6, VAPOR DIFFUSION, HANGING DROP, TEMPERATURE 273K, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.97935 Å / Beamline: 19-ID / Wavelength: 0.97935 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 2, 2007 / Details: MIRROR |
| Radiation | Monochromator: SI 111 CHANNEL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97935 Å / Relative weight: 1 |
| Reflection | Av R equivalents: 0.162 / Number: 125076 |
| Reflection | Resolution: 2.5→50 Å / Num. all: 125076 / Num. obs: 125076 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 10.7 % / Biso Wilson estimate: 44.2 Å2 / Rmerge(I) obs: 0.162 / Rsym value: 0.162 / Net I/σ(I): 21.875 |
| Reflection shell | Resolution: 2.5→2.59 Å / Redundancy: 9.7 % / Rmerge(I) obs: 0.746 / Mean I/σ(I) obs: 2.694 / Rsym value: 0.746 / % possible all: 99.6 |
| Cell measurement | Reflection used: 125076 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2o8n Resolution: 2.5→50 Å / Cor.coef. Fo:Fc: 0.954 / Cor.coef. Fo:Fc free: 0.93 / Occupancy max: 1 / Occupancy min: 1 / SU B: 16.393 / SU ML: 0.173 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.237 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS; U VALUES: RESIDUAL ONLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 96.97 Å2 / Biso mean: 45.999 Å2 / Biso min: 22.88 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→50 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.504→2.569 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
