| 登録情報 | データベース: PDB / ID: 3nhx
|
|---|
| タイトル | Crystal Structure of Ketosteroid Isomerase D99N from Pseudomonas Testosteroni (tKSI) with 4-Androstene-3,17-dione Bound |
|---|
 要素 要素 | Steroid Delta-isomerase |
|---|
 キーワード キーワード | ISOMERASE |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
steroid Delta-isomerase / steroid Delta-isomerase activity / steroid metabolic process類似検索 - 分子機能 Steroid delta5-4-isomerase / Ketosteroid isomerase / Nuclear transport factor 2 (NTF2) domain / SnoaL-like domain / SnoaL-like domain / Nuclear Transport Factor 2; Chain: A, - #50 / NTF2-like domain superfamily / Nuclear Transport Factor 2; Chain: A, / Roll / Alpha Beta類似検索 - ドメイン・相同性 4-ANDROSTENE-3-17-DIONE / Steroid Delta-isomerase類似検索 - 構成要素 |
|---|
| 生物種 |  Comamonas testosteroni (バクテリア) Comamonas testosteroni (バクテリア) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.59 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.59 Å |
|---|
 データ登録者 データ登録者 | Gonzalez, A. / Tsai, Y. / Schwans, J. / Ruben, E. / Sunden, F. / Herschlag, D. |
|---|
 引用 引用 | ジャーナル: To be Published
タイトル: Crystal Structure of Ketosteroid Isomerase D99N from Pseudomonas Testosteroni (tKSI) with 4-Androstene-3,17-dione Bound
著者: Schwans, J. / Ruben, E. / Sunden, F. / Gonzalez, A. / Tsai, Y. / Herschlag, D. |
|---|
| 履歴 | | 登録 | 2010年6月14日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2011年11月23日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2023年9月6日 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / struct_ref_seq_dif / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード














 PDBj
PDBj

 集合体
集合体

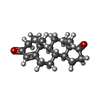
 分子量: 286.409 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C19H26O2
分子量: 286.409 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C19H26O2
 分子量: 96.063 Da / 分子数: 10 / 由来タイプ: 合成 / 式: SO4
分子量: 96.063 Da / 分子数: 10 / 由来タイプ: 合成 / 式: SO4 分子量: 18.015 Da / 分子数: 143 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 143 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製
 解析
解析