Resolution: 1.7→19.983 Å / Cor.coef. Fo:Fc: 0.957 / Cor.coef. Fo:Fc free: 0.933 / SU B: 1.828 / SU ML: 0.062 / Cross valid method: THROUGHOUT / ESU R: 0.103 / ESU R Free: 0.103 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.1985
2932
5.057 %
RANDOM
Rwork
0.1597
55045
-
-
all
0.162
-
-
-
obs
-
57977
97.794 %
-
Solvent computation
Ion probe radii: 1 Å / Shrinkage radii: 1 Å / VDW probe radii: 1.4 Å / Solvent model: MASK BULK SOLVENT
Displacement parameters
Biso mean: 14.205 Å2
Baniso -1
Baniso -2
Baniso -3
1-
0.002 Å2
0 Å2
-0.002 Å2
2-
-
0.001 Å2
0 Å2
3-
-
-
-0.002 Å2
Refinement step
Cycle: LAST / Resolution: 1.7→19.983 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
4086
0
6
726
4818
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.013
0.017
4242
X-RAY DIFFRACTION
r_bond_other_d
0.001
0.016
4023
X-RAY DIFFRACTION
r_angle_refined_deg
1.141
1.807
5747
X-RAY DIFFRACTION
r_angle_other_deg
0.443
1.571
9301
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
6.408
5.146
549
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
11.537
10
733
X-RAY DIFFRACTION
r_dihedral_angle_6_deg
16.829
10
179
X-RAY DIFFRACTION
r_chiral_restr
0.061
0.2
642
X-RAY DIFFRACTION
r_gen_planes_refined
0.005
0.02
4854
X-RAY DIFFRACTION
r_gen_planes_other
0.001
0.02
922
X-RAY DIFFRACTION
r_nbd_refined
0.205
0.2
799
X-RAY DIFFRACTION
r_symmetry_nbd_other
0.172
0.2
3601
X-RAY DIFFRACTION
r_nbtor_refined
0.172
0.2
2110
X-RAY DIFFRACTION
r_symmetry_nbtor_other
0.075
0.2
2110
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.132
0.2
483
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_other
0.059
0.2
2
X-RAY DIFFRACTION
r_symmetry_nbd_refined
0.106
0.2
1
X-RAY DIFFRACTION
r_nbd_other
0.129
0.2
35
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_refined
0.139
0.2
25
X-RAY DIFFRACTION
r_mcbond_it
1.169
1.12
2099
X-RAY DIFFRACTION
r_mcbond_other
1.164
1.121
2099
X-RAY DIFFRACTION
r_mcangle_it
1.78
2.009
2622
X-RAY DIFFRACTION
r_mcangle_other
1.788
2.009
2623
X-RAY DIFFRACTION
r_scbond_it
2.324
1.402
2143
X-RAY DIFFRACTION
r_scbond_other
2.323
1.403
2141
X-RAY DIFFRACTION
r_scangle_it
3.583
2.428
3118
X-RAY DIFFRACTION
r_scangle_other
3.582
2.43
3119
X-RAY DIFFRACTION
r_lrange_it
5.233
13.429
4996
X-RAY DIFFRACTION
r_lrange_other
5.061
12.73
4885
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Rfactor all
Num. reflection all
Fsc free
Fsc work
% reflection obs (%)
WRfactor Rwork
1.7-1.744
0.234
196
0.185
3367
0.188
4337
0.972
0.982
82.1536
0.158
1.744-1.791
0.224
219
0.179
3700
0.182
4252
0.968
0.983
92.1684
0.152
1.791-1.843
0.232
187
0.178
3865
0.181
4101
0.967
0.982
98.8052
0.152
1.843-1.899
0.186
195
0.164
3768
0.166
3972
0.977
0.983
99.7734
0.143
1.899-1.961
0.209
179
0.162
3692
0.164
3878
0.972
0.983
99.8195
0.142
1.961-2.029
0.187
173
0.158
3551
0.159
3730
0.978
0.984
99.8391
0.142
2.029-2.104
0.207
168
0.158
3484
0.16
3662
0.973
0.985
99.7269
0.144
2.104-2.189
0.183
209
0.149
3269
0.151
3484
0.978
0.986
99.8278
0.141
2.189-2.286
0.202
163
0.164
3182
0.166
3350
0.975
0.984
99.8507
0.154
2.286-2.396
0.165
159
0.147
3010
0.148
3175
0.981
0.987
99.811
0.139
2.396-2.523
0.19
156
0.155
2903
0.157
3064
0.975
0.985
99.8368
0.147
2.523-2.673
0.207
175
0.146
2692
0.15
2871
0.975
0.987
99.8607
0.143
2.673-2.854
0.198
139
0.146
2568
0.149
2716
0.974
0.986
99.6686
0.144
2.854-3.078
0.203
125
0.162
2414
0.164
2544
0.973
0.984
99.8035
0.165
3.078-3.364
0.19
112
0.159
2265
0.161
2383
0.978
0.985
99.7482
0.167
3.364-3.747
0.203
115
0.154
1987
0.157
2113
0.976
0.985
99.4794
0.165
3.747-4.302
0.193
84
0.148
1796
0.15
1885
0.98
0.986
99.7347
0.162
4.302-5.209
0.194
80
0.158
1538
0.16
1628
0.985
0.988
99.3857
0.18
5.209-7.128
0.243
60
0.202
1228
0.204
1290
0.975
0.982
99.845
0.221
7.128-19.983
0.154
38
0.179
766
0.177
806
0.983
0.985
99.7519
0.203
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Rickettsia prowazekii (bacteria)
Rickettsia prowazekii (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj Assembly
Assembly



 Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na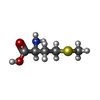
 Type: L-peptide linking / Mass: 149.211 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H11NO2S
Type: L-peptide linking / Mass: 149.211 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H11NO2S
 Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe / Feature type: SUBJECT OF INVESTIGATION
Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 726 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 726 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing