 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3mms: Crystal structure of Streptococcus pneumoniae MTA/SAH nucleosidas... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3mms | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Streptococcus pneumoniae MTA/SAH nucleosidase in complex with 8-aminoadenine | ||||||
 Components Components | 5'-methylthioadenosine / S-adenosylhomocysteine nucleosidase | ||||||
 Keywords Keywords | HYDROLASE / mixed alpha/beta hydrolase | ||||||
| Function / homology |  Function and homology information Function and homology informationadenosylhomocysteine nucleosidase / adenosylhomocysteine nucleosidase activity / methylthioadenosine nucleosidase activity / : / nucleoside catabolic process / : / cytosol Similarity search - Function | ||||||
| Biological species |   Streptococcus pneumoniae (bacteria) Streptococcus pneumoniae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.6 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.6 Å | ||||||
 Authors Authors | Siu, K.K.W. / Lee, J.E. / Horvatin-Mrakovcic, C. / Howell, P.L. | ||||||
 Citation Citation | Journal: TO BE PUBLISHED Title: Crystal structure of Streptococcus pneumoniae MTA/SAH nucleosidase in complex with 8-aminoadenine Authors: Siu, K.K.W. / Lee, J.E. / Horvatin-Mrakovcic, C. / Howell, P.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3mms.cif.gz | 61.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3mms.ent.gz | 45.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3mms.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mm/3mmsftp://data.pdbj.org/pub/pdb/validation_reports/mm/3mms | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 24651.988 Da / Num. of mol.: 1 / Mutation: T23A, A39V, D64V, T184A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus pneumoniae (bacteria) / Strain: ATCC 6303 / Gene: mtnN, pfs, SPP_0997, spr0894 / Plasmid: pET28a / Production host: References: UniProt: Q8DQ16, UniProt: A0A0H2UPP7*PLUS, methylthioadenosine nucleosidase | ||||
|---|---|---|---|---|---|
| #2: Chemical | 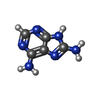  Mass: 150.141 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H6N6 Mass: 150.141 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H6N6#3: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 213 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 213 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.61 Å3/Da / Density % sol: 52.96 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 1.26 M sodium citrate, 90 mM sodium HEPES, pH 7.5, 10 % (v/v) glycerol, vapor diffusion, hanging drop, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.54 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Apr 16, 2002 |
| Radiation | Monochromator: CONFOCAL MULTILAYER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→32.58 Å / Num. obs: 34861 / % possible obs: 99.7 % / Redundancy: 17.28 % / Rmerge(I) obs: 0.041 / Χ2: 1.21 / Net I/σ(I): 13.6 / Scaling rejects: 12528 |
| Reflection shell | Resolution: 1.6→1.66 Å / Rmerge(I) obs: 0.241 / Mean I/σ(I) obs: 2.8 / % possible all: 98.1 |
-Phasing
| Phasing | Method: molecular replacement |
|---|---|
| Phasing MR | Method translation: &STRIP%trans_method |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.6→35 Å / Occupancy max: 1 / Occupancy min: 0 / σ(F): 0
| ||||||||||||||||||||||||||||
| Solvent computation | Bsol: 46.078 Å2 | ||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 55.21 Å2 / Biso mean: 16.497 Å2 / Biso min: 4.57 Å2
| ||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→35 Å
| ||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||
| Xplor file |
|
