- PDB-3jvu: Crystal structure of unliganded P. aeruginosa PilT -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 3jvu
Title
Crystal structure of unliganded P. aeruginosa PilT
Components
Twitching mobility protein
Keywords
ATP binding protein / Motor protein / P-loop atpase / type iv pili / ATP-binding / Fimbrium / Nucleotide-binding / Transport
Function / homology
Function and homology information
pilus retraction / type IV pilus / type IV pilus-dependent motility / ATP hydrolysis activity / ATP binding / cytosol Similarity search - Function
Beta-Lactamase - #90 / Pilus retraction protein PilT/PilU / Bacterial type II secretion system protein E signature. / : / Type II/IV secretion system protein / Type II/IV secretion system protein / Beta-Lactamase / P-loop containing nucleotide triphosphate hydrolases / ATPases associated with a variety of cellular activities / AAA+ ATPase domain ...Beta-Lactamase - #90 / Pilus retraction protein PilT/PilU / Bacterial type II secretion system protein E signature. / : / Type II/IV secretion system protein / Type II/IV secretion system protein / Beta-Lactamase / P-loop containing nucleotide triphosphate hydrolases / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / Rossmann fold / P-loop containing nucleoside triphosphate hydrolase / 2-Layer Sandwich / 3-Layer(aba) Sandwich / Alpha Beta Similarity search - Domain/homology
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Pseudomonas aeruginosa (bacteria)
Pseudomonas aeruginosa (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj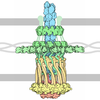
 Assembly
Assembly


 Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl
 Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7
Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 18.015 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 21-ID-D / Wavelength: 0.979 Å
/ Beamline: 21-ID-D / Wavelength: 0.979 Å Processing
Processing