 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3huf | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the S. pombe Nbs1-Ctp1 complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | CELL CYCLE / Nbs1 / FHA domain / BRCT domain / phosphoprotein binding / phosphoserine binding / DNA repair / Ctp1 / Chromosomal protein / DNA damage / Nucleus / Phosphoprotein / Telomere / Meiosis | ||||||
| Function / homology |  Function and homology information Function and homology informationdouble-strand/single-strand DNA junction binding / DNA-DNA tethering activity / endodeoxyribonuclease activator activity / stalled replication fork localization to nuclear periphery / HDR through MMEJ (alt-NHEJ) / meiotic DNA double-strand break processing / gene conversion at mating-type locus / DNA Damage/Telomere Stress Induced Senescence / Sensing of DNA Double Strand Breaks / : ...double-strand/single-strand DNA junction binding / DNA-DNA tethering activity / endodeoxyribonuclease activator activity / stalled replication fork localization to nuclear periphery / HDR through MMEJ (alt-NHEJ) / meiotic DNA double-strand break processing / gene conversion at mating-type locus / DNA Damage/Telomere Stress Induced Senescence / Sensing of DNA Double Strand Breaks / : / Recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double strand breaks / flap-structured DNA binding / Mre11 complex / chromosome, telomeric repeat region / mitotic recombination-dependent replication fork processing / DNA end binding / phosphorylation-dependent protein binding / Y-form DNA binding / double-strand break repair involved in meiotic recombination / DNA double-strand break processing / bubble DNA binding / mitotic DNA replication checkpoint signaling / telomere maintenance via recombination / chromatin-protein adaptor activity / mitotic intra-S DNA damage checkpoint signaling / mitotic G2 DNA damage checkpoint signaling / replication fork processing / telomere maintenance / double-strand break repair via homologous recombination / double-strand break repair via nonhomologous end joining / double-strand break repair / single-stranded DNA binding / site of double-strand break / double-stranded DNA binding / endonuclease activity / molecular adaptor activity / Hydrolases; Acting on ester bonds / damaged DNA binding / DNA repair / hydrolase activity / identical protein binding / nucleus Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.15 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.15 Å | ||||||
 Authors Authors | Williams, R.S. / Guenther, G. / Tainer, J.A. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2009 Title: Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Authors: Williams, R.S. / Dodson, G.E. / Limbo, O. / Yamada, Y. / Williams, J.S. / Guenther, G. / Classen, S. / Glover, J.N. / Iwasaki, H. / Russell, P. / Tainer, J.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3huf.cif.gz | 198.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3huf.ent.gz | 159 KB | Display | PDB format |
| PDBx/mmJSON format | 3huf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hu/3hufftp://data.pdbj.org/pub/pdb/validation_reports/hu/3huf | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| 5 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 36744.961 Da / Num. of mol.: 3 Fragment: N-terminal FHA-BRCT1-BRCT2 domains (residues 1-321) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: nbs1, SPBC6B1.09c / Production host:  #2: Protein/peptide | | Mass: 1667.467 Da / Num. of mol.: 1 / Fragment: SXT sites (residues 72-84) / Source method: obtained synthetically / Source: (synth.) #3: Chemical | 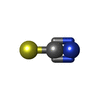  Mass: 58.082 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CNS Mass: 58.082 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CNS#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 428 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 428 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.77 Å3/Da / Density % sol: 55.57 % |
|---|---|
| Crystal grow | Temperature: 281 K Details: 18% polyethylene glycol 3350, 100 mM MES pH 6.1-6.5, 260 mM potassium thiocyanate, VAPOR DIFFUSION, HANGING DROP, temperature 281K PH range: 6.1-6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 12.3.1 / Wavelength: 1.1158 / Beamline: 12.3.1 / Wavelength: 1.1158 |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 1, 2008 |
| Radiation | Monochromator: SI 111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1158 Å / Relative weight: 1 |
| Reflection | Resolution: 2.15→50 Å / Num. obs: 66677 / % possible obs: 97.1 % / Observed criterion σ(I): 2.3 / Redundancy: 3.4 % / Rmerge(I) obs: 0.047 / Net I/σ(I): 23 |
| Reflection shell | Resolution: 2.15→2.23 Å / Redundancy: 2.6 % / Rmerge(I) obs: 0.433 / Mean I/σ(I) obs: 2.3 / % possible all: 86.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.15→50 Å / Cor.coef. Fo:Fc: 0.942 / Cor.coef. Fo:Fc free: 0.921 / SU B: 10.54 / SU ML: 0.144 / TLS residual ADP flag: LIKELY RESIDUAL / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / ESU R: 0.232 / ESU R Free: 0.202 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 44.65 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.15→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.15→2.21 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
